

1. Introduction
Oral squamous cell carcinoma accounts for approximately 90% of all oral malignancies and is one of the most prevalent forms of oral cancer in South Asia (Chai et al., 2020). High incidence and mortality rates have been observed in Asia compared to other regions, and it mostly affects middle-aged and elderly men (Bugshan & Farooq, 2020). OSCC arises from squamous epithelial cells due to uncontrolled growth and affects the mouth, lips, tongue, and other regions of the oral cavity (Miguelanez-Medran et al., 2019). The oral cavity and oropharynx are the common sites affected in OSCC (Bugshan & Farooq, 2020). The significant risk factors for OSCC are linked to various habits, such as tobacco smoking, alcohol intake, betel nut chewing, human papillomavirus infection, nutritional deficiency, immune deficiency, and other lifestyle habits (Choi et al., 2025; Zhao et al., 2024). These factors contribute to OSCC through chronic inflammation in cells, DNA damage, and genetic modifications (M. Li et al., 2025). Smoking aids in tumor progression by suppressing immunity in the oral region and increases the risk of carcinogenesis by affecting tumor suppressor genes, such as p53 and PTEN (Tan et al., 2023). Cigarettes contain various harmful substances, including nitrosamines, aromatic amines, and polycyclic aromatic hydrocarbons (PAHs), which increase the risk of OSCC (Imbesi Bellantoni et al., 2023). Another common cause is alcohol consumption, and the synergistic effects of alcohol use and smoking further increase the risk (Feller et al., 2013). Likewise, betel nut chewing, along with tobacco, is more carcinogenic (approximately 15 times) than without smoking. It releases reactive oxygen species (ROS), which cause toxicity, genetic alterations, and initiate tumorigenesis (P.-H. Chen et al., 2017). Compounds such as arecoline and arecaidine from betel nut can trigger DNA damage and induce OSCC (Y.-C. Li et al., 2019). Apart from these causes, genetic alterations and epigenetic modifications also contribute to OSCC development (Tan et al., 2023). At present, treatment methods such as chemotherapy, radiotherapy, and surgical procedures are used to treat oral cancers (Kijowska et al., 2024). Despite several treatment methods, patient outcomes in OSCC are compromised due to the development of resistance to chemotherapeutic agents, including cisplatin and 5-fluorouracil. Moreover, patients experience severe side effects during treatment procedures and may experience recurrence. Notably, this type of cancer is often diagnosed at later stages, where treatment becomes more difficult, leading to poor survival rates (Gormley, 2022; Menditti et al., 2023).
Plant-based medicines have been used for many centuries in various traditional medicine systems as an alternative to synthetic drugs. These plant-based products offer several advantages to the human body, making them a more affordable option for multiple treatments, including cancer (Ramsridhar et al., 2025). Natural compounds from various plants have shown promising therapeutic potential for use in cancer therapy due to their antitumor properties. They contain multiple kinds of active phytochemicals, such as alkaloids, terpenoids, flavonoids, and phenolic compounds (Lichota & Gwozdzinski, 2018). These compounds exhibit no or low toxicity, are readily available, and are low cost, which makes them potential candidates in cancer treatment (Yu et al., 2022). Additionally, these compounds can inhibit cell growth and induce apoptosis by targeting various cancer-related pathways involved in tumor development and metastasis (Choudhari et al., 2020).
Syzygium cumini is a member of the Myrtaceae family, commonly known as Jamun, Indian blackberry, Java plum, or Malabar plum. It is widely found in tropical and subtropical regions, with its origin in India and Indonesia (Das et al., 2023). Various parts of S. cumini, including fruits, seeds, bark, and leaves, possess significant medicinal value and have been used for centuries in various traditional medicine systems such as Siddha, Ayurveda, Unani, Homeopathy, and Chinese medicine to treat various diseases (L. Li et al., 2021). These components contain various active phytochemicals, including flavonoids, anthocyanins, phenols, and terpenoids, which exert antioxidant, anti-inflammatory, antimicrobial, anti- diabetic, antiviral, and anticancer properties (Chhikara et al., 2018; Sawant et al., 2015). The fruits are purple to blackish in color due to high anthocyanin content (Gajera et al., 2018). In addition to anthocyanins, the fruits contain condensed and hydrolysable tannins (such as ellagitannins and gallotannins), predominantly found in the skin, which are a natural source of antioxidants that exhibit strong antioxidant capabilities (Das et al., 2023; Tavares et al., 2016). The fruit has been used to treat various physiological illnesses such as dysentery, gastric issues, and other digestive problems. These are rich in fiber, vitamins, and minerals such as calcium, potassium, and iron (Qamar et al., 2022). Additionally, jamun fruits are a good source of carbohydrates such as glucose, fructose, sucrose, and maltose, as well as amino acids such as alanine, asparagine, and tyrosine (Al-Dhabi & Ponmurugan, 2020; Aqil et al., 2012). Consuming this fruit offers several health benefits, and it has been used for the management of diabetes since ancient times in traditional medicine (Qamar et al., 2022). It lowers blood sugar levels in diabetic patients and also helps in purifying the blood. Due to the presence of iron, it boosts hemoglobin levels and acts as a blood-purifying agent (El-Safy et al., 2023). S. cumini, being a rich reservoir of various bioactive phytoconstituents, has gained significant interest in the field of pharmacology and ethnomedicine (Ravi Ahirwar*, 2025).
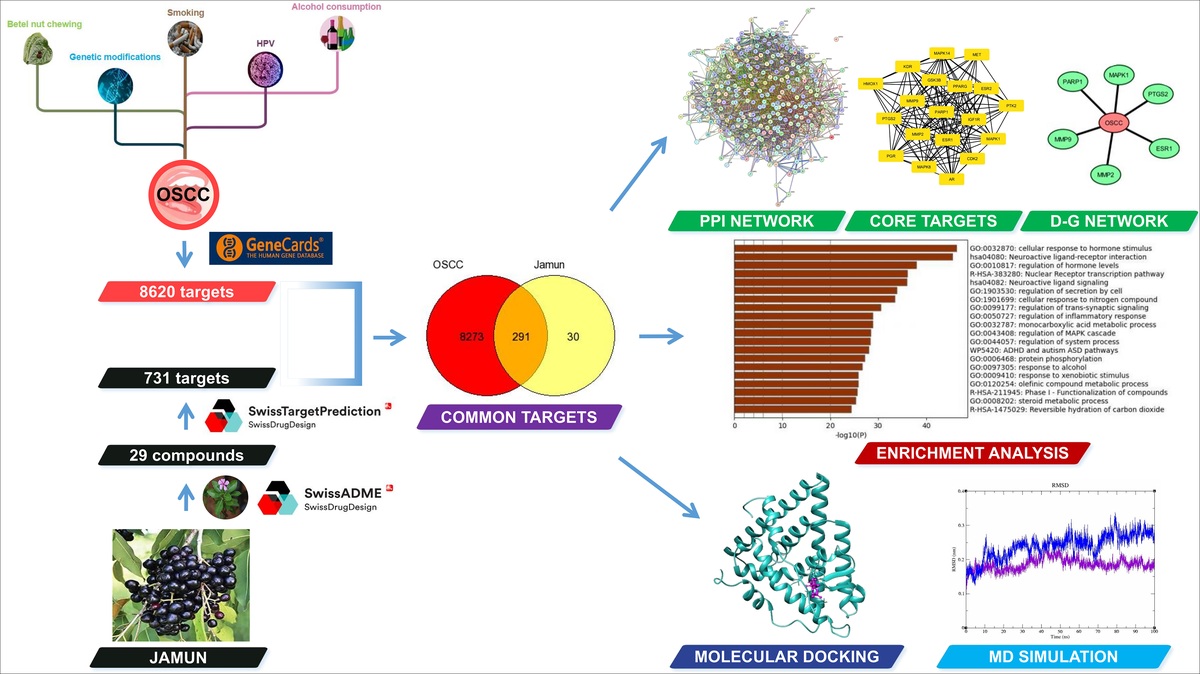
Recently, the network pharmacology approach has been extensively utilized in drug discovery to unveil the therapeutic mechanisms of herbal- and traditional-based medicines in the treatment of various diseases (S. Yang et al., 2024). This approach has shifted the paradigm from the traditional single-target, single-drug process to a multi-target, multi-drug concept in which drugs act on multiple targets (Zhai et al., 2021). Network pharmacology integrates bioinformatics and systems biology to analyze complex compound-target interactions involved in disease treatment (Sakuludomkan et al., 2025). This work aims to identify the active phytochemical compounds of Syzygium cumini and elucidate their mechanism of action for the treatment of OSCC. A network pharmacology-based approach was employed, integrating molecular docking and MD simulation analysis to explore the therapeutic potential of S. cumini. This approach helped identify key phytochemical compounds, potential molecular targets, and underlying pathways involved in the pathogenesis of OSCC. The graphical abstract is shown in Figure 1.
2. Materials and Methods
2.1. Conceptual Design, Study Hypothesis, and Data retrieval
This work is an in silico study conducted between June 2025 and October 2025, utilizing publicly available datasets, bioinformatics analyses, and molecular modelling workflows. The study was designed as a hypothesis-driven in silico investigation to elucidate the potential molecular mechanisms by which phytochemicals derived from Syzygium cumini may exert therapeutic effects against OSCC, using an integrative approach. This study did not involve human participants, patient data, or animals and did not require ethics committee approval or informed consent. The bioactive compounds present in Syzygium cumini were obtained using the IMPPAT database (Mohanraj et al., 2018). The botanical name of the Jamun fruit, “Syzygium cumini”, was used as the keyword to search for the phytochemicals. The phytochemical compounds present in the fruit were collected and considered for this study.
2.2. Screening of Bioactive Compounds and Target Identification
The obtained active compounds were subjected to ADME analysis using SwissADME to predict their drug-likeness and oral bioavailability, which are crucial in determining the characteristics of drugs (Daina et al., 2017). Compounds that did not satisfy these criteria were excluded from further studies. The potential targets of the selected compounds of Syzygium cumini were predicted using SwissTargetPrediction (Daina et al., 2019). The SMILES of each phytochemical was used as the query to retrieve targets relevant to Homo sapiens with a probability score of 1 and were taken for subsequent analysis.
2.3. Prediction of OSCC-related target genes
Collecting disease-related genes is essential for understanding the mechanisms of herbal formulations or plant-derived products and their active phytochemicals in disease therapy. The genes associated with OSCC were collected from the GeneCards database using the keyword “oral squamous cell carcinoma” (Stelzer et al., 2016)
2.4. Construction of a Venn diagram
The potential common targets associated with OSCC and the active phytochemicals in jamun fruit were identified using the GeneVenn tool. This tool generated a Venn diagram comprising potential overlapping targets of OSCC and the jamun fruit. The UniProt ID of the protein targets was given as input in the Venn diagram tool.
2.5. Gene Ontology (GO) and Pathway Mapping of Common Targets
The potential overlapping targets were subjected to GO and KEGG enrichment analyses using the Metascape tool (Zhou et al., 2019). The UniProt ID of the protein targets was given as the input, and the species was selected as Homo sapiens. Metascape is a curated tool that analyses gene sets by integrating various ontology sources such as GO Biological Processes, KEGG pathways, and Reactome Gene Sets. The p-values and q-values in the Metascape tool were calculated based on the cumulative hypergeometric distribution and Benjamini–Hochberg, which help in multiple testing, thereby improving the reliability of enrichment results. Terms with p-value of < 0.01, q-value of < 0.05, and an enrichment factor > 1.5 were considered statistically significant.
2.6. PPI Network Analysis and Hub Target Extraction
The UniProt ID of protein targets was submitted to the STRING database to build a protein–protein interaction (PPI) network with a threshold of 0.4 (medium confidence) (Szklarczyk et al., 2023). The output file was downloaded from STRING and imported into Cytoscape software for better visualization and further analysis (Shannon et al., 2003). The MCODE plugin was used to identify the highly connected PPI sub-network or clustered targets within the network by adjusting specific parameters—node score cutoff (0.2), K-Core cutoff (2), maximum depth (100), and degree cutoff (2). DisGeNET, another plugin, was used to identify gene–disease associations related to OSCC.
2.7. Molecular Docking
Molecular docking was performed for the identified protein targets with their corresponding phytoconstituents using AutoDock Tools v1.5.6 (Morris et al., 2009). The proteins were prepared by adding necessary charges and converted into PDBQT format. Similarly, all ligands were converted into the same format, and docking was carried out. The binding sites were defined for every target protein, and the grid box was generated accordingly. The default parameters of the Lamarckian Genetic Algorithm were utilized to perform docking, and the binding affinities between each protein and ligand were calculated. BIOVIA Discovery Studio v24.1.0.23298 was employed to visualize the interactions within the protein–ligand complexes (Dassault Systèmes BIOVIA, n.d.).
2.8. MD simulations
Based on molecular docking, the top three protein–ligand complexes with the highest binding affinities were selected for simulation using the GROMACS 2023.2 package (Abraham et al., 2023). The protein parameterization was performed using the CHARMM27 force field, while the ligand topology was generated through the SwissParam webserver (Zoete et al., 2011). The complexes were converted into GROMACS format, and the hydrogen atoms were removed. The TIP3P water model was utilized to solvate the complexes. Subsequently, neutralization of the system was achieved by adding appropriate counter ions. Following this, an energy minimization step was performed using the steepest descent integrator to eliminate steric clashes and hindrances within the complexes. All systems were subjected to equilibration, which was carried out in two steps, namely, the NVT and the NPT ensembles. During NVT and NPT equilibration, the Berendsen thermostat (Berendsen et al., 1984) was used to maintain constant volume for 100 ps and a constant temperature of 310 K, while the Parrinello–Rahman barostat (Parrinello & Rahman, 1981) was used to maintain a pressure of 1 bar for 100 ps, respectively. A production run of 100 ns was performed after the equilibration phases, and the output trajectory files were analyzed to compute root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), hydrogen bond (H-bond), and solvent accessible surface area (SASA).
3. Results
3.1. Prediction of Bioactive Compounds
Using the IMPPAT database, we obtained a total of 32 active compounds that were present in the Syzygium cumini (fruit) and are shown in Table S1.
3.2. Screening of Bioactive Compounds and Target Identification
Using SwissADME, the pharmacokinetic properties (Absorption, Distribution, Metabolism, and Excretion) of the compounds were predicted, and the compounds that satisfied Lipinski’s rule of five were screened (zero and one violation) (Table S2). A total of 29 compounds were retrieved, as only three compounds violated the rule of five and were excluded. The potential human targets for the screened compounds were obtained using SwissTargetPrediction. We obtained 731 human targets with a probability value of 1 for the 29 screened compounds related to OSCC (Supplementary File 1).
3.3. Prediction of OSCC-related target genes
A total of 9,873 target genes related to OSCC were retrieved from the GeneCards database, out of which 8620 are protein-coding genes and were selected and considered for further analysis (Supplementary File 2).
3.4. Construction of a Venn diagram
The common targets that overlap between OSCC-related targets and the targets of S. cumini were identified using a Venn diagram. The intersection of these targets resulted in 29 overlapping targets and was considered a key target that might play a major role in OSCC (Figure S1).
3.5. Gene Ontology (GO) and Pathway Mapping of Common Targets
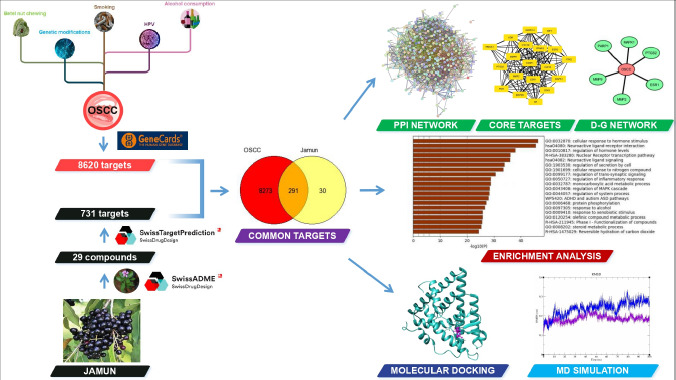
A total of 290 overlapping targets were subjected to Gene Ontology (GO) and pathway enrichment analyses using the Metascape tool. More than 50 genes were associated with biological processes (BP), including cellular response to hormone stimulus (GO:0032870), regulation of hormone levels (GO:0010817), regulation of secretion by cell (GO:1903530), cellular response to nitrogen compound (GO:1901699), and regulation of MAPK cascade (GO:0043408), which emerged as the most significantly enriched terms. Additionally, other processes such as regulation of trans-synaptic signaling (GO:0099177), regulation of inflammatory response (GO:0050727), response to alcohol (GO:0097305), and protein phosphorylation (GO:0006468) were also significantly enriched for BP. KEGG pathway enrichment analysis revealed that neuroactive ligand–receptor interaction and neuroactive ligand signaling were the most significantly enriched pathways, with 18.3% and 12.8% of the genes involved, respectively. Additionally, the Nuclear Receptor transcription pathway, Phase I – Functionalization of compounds, and Reversible hydration of carbon dioxide were identified from Reactome pathway enrichment, which correspond to 4.14% and 8.62% of genes (Figure 2).
3.6. PPI Network Analysis and Hub Target Extraction
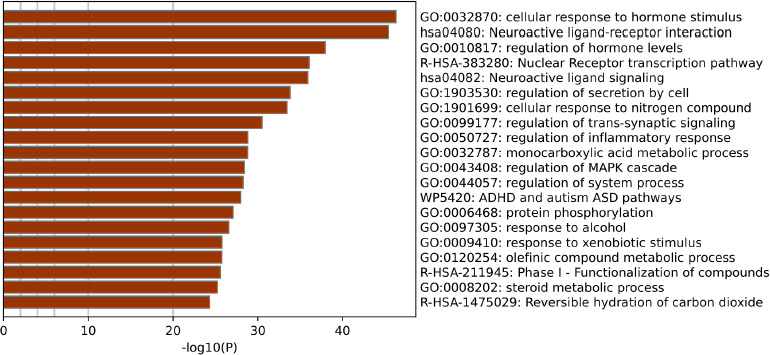
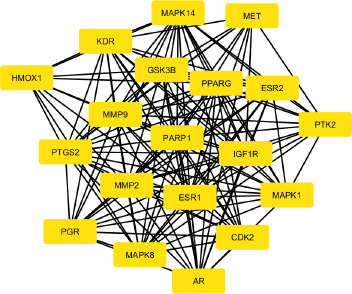
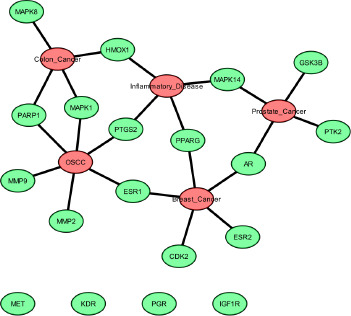
The protein–protein interaction (PPI) network was built using STRING (Figure S2), and the output file was downloaded in tsv format, which is compatible with Cytoscape. The PPI network contains 94 nodes and 2687 edges, with an average node degree of 18.7 and an average local clustering coefficient of 0.45. Subsequent analyses were performed using Cytoscape software. Using the MCODE plugin, we identified a total of 19 core targets, namely Mitogen-activated protein kinase 14 (MAPK14), hepatocyte growth factor receptor (MET), Vascular endothelial growth factor receptor 2 (KDR), glycogen synthase kinase-3 beta (GSK3B), Peroxisome proliferator-activated receptor gamma (PPARG), Estrogen receptor beta (ESR2), protein tyrosine kinase 2 (PTK2), insulin-like growth factor I receptor (IGF1R), Mitogen-activated protein kinase 1 (MAPK1), Cyclin-dependent kinase 2 (CDK2), Androgen receptor (AR), Mitogen-activated protein kinase 8 (MAPK8), Progesterone receptor (PGR), matrix metalloproteinase 2 (MMP2), poly [ADP-ribose] polymerase-1 (PARP1), matrix metalloproteinase 9 (MMP9), Prostaglandin G/H synthase 2 (PTGS2), Heme oxygenase 1 (HMOX1), and Estrogen receptor (ESR1) (Figure 3). To determine the relevance to OSCC, we performed gene–disease association analysis using DisGeNET and identified several targets within the obtained core target hub. These include MAPK1, PTGS2, ESR1, MMP2, MMP9, and PARP1 (Figure 4). These targets were associated with OSCC, indicating their potential involvement in disease pathogenesis and were considered for molecular docking.
3.7. Molecular Docking
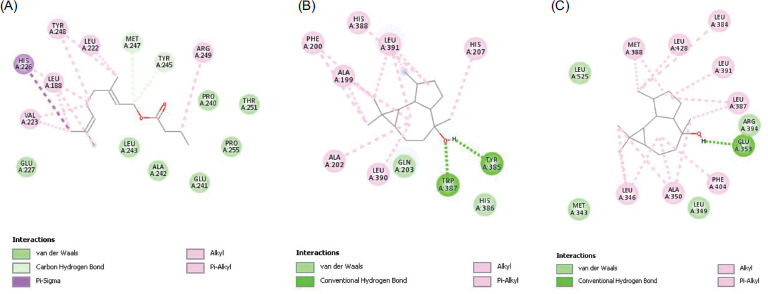
Based on the PPI network and hub target analysis using STRING, Cytoscape, and DisGeNET, six OSCC-related targets were selected: MAPK1, PTGS2, ESR1, MMP2, MMP9, and PARP1, and molecular docking was performed for these targets with their corresponding phytochemicals. Among all active phytochemicals, Geranyl butyrate exhibited the highest binding energy of -9.04 kcal/mol with MMP9. Similarly, Ledol exhibited the second-highest binding energy of -8.98 kcal/mol with PTGS2. Following this, (-)-Globulol exhibited the third-highest binding energy of -8.55 kcal/mol with ESR1. The binding energies of all the phytochemicals and protein targets are shown in Table 1, and the interactions of the top three protein–ligand complexes are shown in Figure 5.
Table 1
Binding energies of OSCC targets with their corresponding Syzygium cumini phytochemicals.
3.8. MD simulations
MD simulations were performed to further evaluate the impact of S. cumini active phytochemicals on OSCC-related targets. The top three protein–ligand complexes, MMP9–Geranyl butyrate, PTGS2–Ledol, and ESR1–(-)-Globulol, which had the highest binding energies, were selected for simulation.
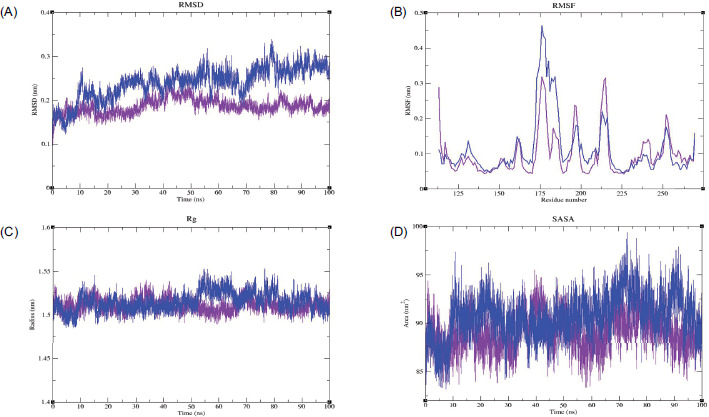
The MMP9–Geranyl butyrate complex exhibited a stable RMSD value with no significant fluctuations throughout the simulation. The complex attained equilibrium after 50 ns, suggesting higher stability due to the binding of Geranyl butyrate. Meanwhile, the MMP9-Apo protein displayed high deviations up to ~ 0.34 nm but reached equilibrium after 80 ns (Figure 6A). RMSF analysis revealed that the MMP9 complex exhibited low flexibility across most residues; however, in certain regions between ~ 180–200 and 209–218, deviations up to ~ 0.25 and 0.3 nm were observed, respectively. In the Apo protein, higher fluctuations were observed compared to the complex, with the highest peak reaching up to ~ 0.47 nm at residues ~ 170–187 (Figure 6B). From the Rg plot, both the ligand-bound and unbound MMP9 exhibited Rg values within the range of ~ 1.48–1.55 nm. The Apo protein maintained its compactness up to ~ 55 ns, after which an increase in the peak up to ~ 1.57 nm was observed. However, the Apo protein converged at the end of the simulation, indicating that it attained a conformational state with high compactness similar to the bound state. On the other hand, the complex did not show any major deviations from the beginning to the end of the simulation, indicating better compactness (Figure 6C). The SASA analysis revealed that the MMP9-Apo structure exhibited more fluctuations compared to the complex, with fluctuations up to ~ 100 nm2 observed throughout the simulation. The MMP9 complex exhibited lower SASA values compared to the Apo form and converged, indicating a reduced solvent-accessible surface area (Figure 6D).
Figure 6
MDS analysis of the MMP9-Geranyl butyrate complex, depicting (A) root mean square deviation (RMSD), (B) root mean square fluctuation (RMSF), (C) radius of gyration (Rg), and (D) solvent-accessible surface area (SASA) profiles, highlighting the structural stability, flexibility, compactness, and solvent exposure of the complex throughout the simulation period. Color scheme: MMP9-Apo is shown in blue, and the MMP9-Geranyl butyrate complex is shown in violet.
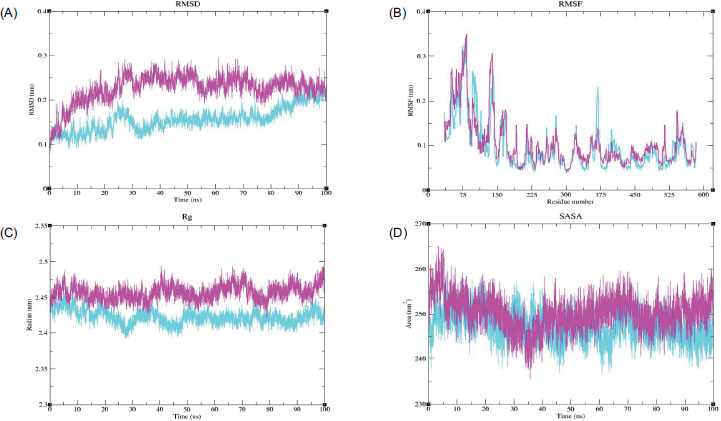
The RMSD plot for PTGS2 showed that the Apo form exhibited higher RMSD values than the PTGS2–Ledol complex. During the initial phase of the simulation, the PTGS2-Apo structure displayed a significant increase in its peaks; however, after ~ 25 ns, the system reached an equilibrium state and maintained an RMSD value between ~ 0.2–0.3 nm. Meanwhile, the complex structure attained its equilibrium state after 30 ns and maintained a stable state up to ~ 80 ns. Towards the end, a slight increase in the trend was observed, reaching up to ~ 0.25 nm, but still lower than the Apo form (Figure 7A). Both systems displayed variable fluctuations and distinct peaks at various regions, which might correspond to loops or unstructured areas. The complex structure indicates a stable conformation with overall lower flexibility in several key regions. In the case of the Apo form, significant peaks were observed at residues 75 (~ 0.35 nm) and 145 (~ 0.31 nm), respectively, indicating that these regions were highly flexible (Figure 7B). In Rg analysis, higher Rg values were observed for the Apo protein, with minimal deviations after ~ 35 ns. The complex structure exhibited relatively lower Rg values than the Apo protein and reached equilibrium around 50 ns, suggesting that the protein maintained its structural compactness throughout the simulation (Figure 7C). Higher SASA values were observed for the PTGS2-Apo system, up to ~ 265 nm2, whereas lower SASA values were observed for the complex, indicating that the protein was tightly packed with fewer solvent-exposed areas (Figure 7D).
Figure 7
MDS analysis of the PTGS2-Ledol complex, depicting (A) root mean square deviation (RMSD), (B) root mean square fluctuation (RMSF), (C) radius of gyration (Rg), and (D) solvent-accessible surface area (SASA) profiles, highlighting the structural stability, flexibility, compactness, and solvent exposure of the complex throughout the simulation period. Color scheme: PTGS2-Apo is shown in magenta, and the PTGS2-Ledol complex is shown in cyan.
The RMSD plot for ESR1 revealed that the Apo protein exhibited constant fluctuations ranging from ~ 0.2–0.3 nm throughout the simulation period. In contrast, the ESR1–(-)-Globulol complex exhibited relatively lower RMSD values than the Apo form, indicating a more stable conformation. After the initial rise, the complex maintained its stability, and convergence was observed at the end (Figure 8A). From the RMSF analysis, a higher fluctuation pattern was observed for the ESR1-Apo protein, with the highest deviations up to ~ 0.5 nm and 0.36 nm at residues 551 and 535, respectively. Meanwhile, the complex demonstrated lower fluctuation patterns across most residues, except at positions 307 (~ 0.5 nm), 465 (~ 0.33 nm), and 578 (~ 0.32 nm), where higher flexibility was observed compared to the Apo form (Figure 8B). The Rg analysis revealed that the complex was more tightly packed, with no significant structural changes observed, confirming its compactness and rigidity. In contrast, the Apo form showed slightly higher Rg values than the complex; however, both systems converged at the end of the simulation (Figure 8C). In the ESR1–(-)-Globulol complex, fluctuations were observed in the initial phase up to ~ 20 ns, after which the system maintained consistently lower SASA values until ~ 70 ns. A sudden peak was observed, indicating slight conformational changes in the protein, after which it converged at the end. In the case of the Apo protein, higher fluctuations were observed, implying that the protein adopted a more open conformation, making it more accessible to the solvent (Figure 8D).
Figure 8
MDS analysis of the ESR1-(-)-Globulol complex, depicting (A) root mean square deviation (RMSD), (B) root mean square fluctuation (RMSF), (C) radius of gyration (Rg), and (D) solvent-accessible surface area (SASA) profiles, highlighting the structural stability, flexibility, compactness, and solvent exposure of the complex throughout the simulation period. Color scheme: ESR1-Apo is shown in turquoise, and the ESR1-(-)-Globulol complex is shown in maroon.
4. Discussion
Oral squamous cell carcinoma is one of the most prevalent cancer types in the head and neck region and is responsible for 90% of the morbidity and mortality (Chamoli et al., 2021). Mainly, smoking, alcohol consumption, and betel nut chewing contribute to OSCC development, and these habits are predominantly observed in countries such as India, Taiwan, and other Southeast Asian countries (Hao et al., 2024). Patients often suffer from various adverse reactions during treatment, which remains a challenge in OSCC (Alqarni et al., 2024). Moreover, the 5-year survival rate of OSCC patients remains lower than 50% due to metastasis and disease recurrence even after treatment, highlighting the need for alternative therapeutic strategies (Cristaldi et al., 2019). Plant-based medicines are now widely used as an alternative or as an adjuvant in cancer therapy due to their minimal or no adverse effects compared to chemotherapeutic drugs (Hashim et al., 2024). This study aimed to investigate the potential pharmacological mechanisms of Syzygium cumini in the treatment of OSCC by employing a network pharmacology approach along with molecular docking and simulation analysis.
The compounds of S. cumini were retrieved and filtered based on Lipinski’s rule of five using the SwissADME tool. Subsequently, human targets for these compounds were identified. On the other hand, OSCC-related genes were collected, and the common overlapping targets associated with OSCC and the targets of S. cumini compounds were identified using a Venn diagram. This significant overlap suggests that multiple targets are commonly involved in both, thus indicating a potential therapeutic relevance of S. cumini in OSCC. Further GO and KEGG functional enrichment analyses revealed various significant biological processes and molecular pathways involved in the pathogenesis of OSCC (Figure 2). The most significant biological processes involved in OSCC include cellular response to hormone stimulus, regulation of hormone levels, regulation of the MAPK cascade, regulation of inflammatory response, and response to alcohol. While OSCC is not hormone dependent, the observed results, such as cellular response to hormone stimulus and regulation of hormone levels, suggest a potential role in OSCC. The MAPK signaling pathways are cascades of three kinases, including MAP kinase kinase kinase (MAPKKK), MAP kinase kinase (MAPKK), and MAP kinase (MAPK). This pathway is primarily involved in cell growth, cell migration, apoptosis, angiogenesis, and metastasis. Deregulation or aberrant activation of this pathway leads to tumorigenesis (Lee et al., 2020; Peng et al., 2017). In any type of cancer, inflammation plays a significant role, including OSCC. Chronic inflammation may lead to the production of cytokines, growth factors, and ROS, which can cause DNA damage, leading to cancer (Singh et al., 2022). Notably, the response to alcohol shows a strong epidemiological association between chronic alcohol consumption and OSCC incidence, as alcohol is linked to approximately 26% of all oral malignancies globally (Tenore et al., 2020).
A PPI network was constructed for the overlapping targets using the STRING database. The STRING network was imported into Cytoscape, and subsequently, the MCODE plugin was utilized to identify the key targets such as MAPK14, ESR2, PTK2, MAPK1, AR, MAPK8, PARP1, MMP9, PTGS2, and ESR1 (Figure 3). Furthermore, DisGeNET was used to perform gene–disease association analysis, and six targets, namely MAPK1, PTGS2, ESR1, MMP2, MMP9, and PARP1, were identified (Figure 4). MAPK1, also known as ERK2, is a member of the mitogen-activated protein kinase (MAPK) signaling pathway, which plays a critical role in regulating various cellular functions, including cell survival, proliferation, apoptosis, and cellular metabolism (Z. Li et al., 2020). Phosphorylation of MAPK1 leads to the activation of downstream signaling pathways and promotes cell migration and proliferation, making it a critical regulatory factor in the progression of OSCC (S. Huang et al., 2024). Overexpression of PTGS2 facilitates angiogenesis and promotes tumor progression by modulating and stabilizing the tumor microenvironment (TME) (Frejborg et al., 2020). It also causes inflammation, prevents apoptosis, and contributes to treatment resistance (Kamal et al., 2024). Tobacco chewing leads to increased expression of PTGS2 and has been detected in oral tissues of affected individuals (R.-Y. Huang & Chen, 2011). There are only a few studies reported for ESR1 in the context of OSCC. In one study, expression of ESR1 was observed, but it was rare in OSCC cases; however, the overall survival rates were decreased in male patients (Doll et al., 2021). Several studies reported the overexpression of MMP2 and MMP9 in OSCCs (Baker et al., 2006; De Vicente et al., 2005; Mashhadiabbas et al., 2012; Mohtasham et al., 2013; Pramanik & Mishra, 2022; Tamamura et al., 2013; Ye et al., 2008). MMP2 and MMP9 are classes of metalloproteinases that play a crucial role in the degradation of the extracellular matrix (ECM) and are associated with tumor invasion and metastasis, including OSCC (Lu et al., 2014; Sanyal et al., 2022). In OSCC, the basement membrane functions as the first barrier of the ECM, separating epithelial and mesenchymal cells, which must be degraded for tumor invasion and metastasis. Both MMP2 and MMP9, in their activated forms, degrade the ECM and type IV collagen, thereby facilitating invasion and metastasis (Kato et al., 2005). Some studies reported that MMP2 is also linked with ERK and/or p38 activity (Chang et al., 2021; W.-E. Yang et al., 2019). The expression of MMP2 has been shown to increase with advanced tumor stage, high histological grade, and tumor invasiveness. It has also been linked with the ERK1/2 signaling pathway, highlighting its importance in OSCC progression (Shrestha et al., 2017). Meanwhile, MMP9 has been associated with the aggressive nature of OSCC (Vilen et al., 2013). In tongue cancer, a subtype of OSCC, elevated MMP9 expression is strongly associated with increased proliferation of tumor cells. The overexpression of MMP9 promotes the degradation of ECM components, predominantly type IV collagen, leading to basement membrane thinning. This process facilitates tumor cell invasion, metastasis, and angiogenesis (Fan et al., 2012). High levels of MMP9 have been found in advanced stages of OSCC and correlate with tumor progression and invasion (Patil et al., 2021). MMP9 can be used as a biomarker to measure metastatic potential in OSCC, and its levels vary according to tumor grade (Patel et al., 2005). In one study, the expression of PARP1 was found to be significantly elevated in patient-derived OSCC tissue samples (Kossatz et al., 2016). In another study, OSCC tumor cells derived from recurrent cases showed significantly higher expression of PARP1. These elevated levels of PARP1 contribute to resistance and recurrence in OSCC (Wang et al., 2022).
The corresponding bioactive compounds of the targets obtained from DisGeNET majorly fall under terpenoids. Terpenoids are natural bioactive compounds with numerous pharmacological benefits, which are made up of isoprene units (Guo et al., 2024). These compounds exhibit various biological properties such as antioxidant, anti-inflammatory, anticancer, and antimicrobial properties (Mitea et al., 2025).
Some terpenoids can inhibit cell differentiation and activate apoptosis, leading to suppression of tumorigenesis. Additionally, these compounds can inhibit angiogenesis and metastasis at advanced stages of cancer (Kamran et al., 2022). Geranyl butyrate, a monoterpenoid, was synthesized from geraniol to determine anticancer activity in vitro, and the study revealed that it demonstrated potent cytotoxic activity against murine leukemia (P388) cells (Widiyarti et al., 2019). A study by Rodríguez et al. demonstrated that Globulol (a sesquiterpenoid) exhibits antioxidant properties and may be utilized as a potential therapeutic agent to neutralize free radicals (Rodriguez et al., 2019). El-Shiekh et al. reported that Globulol had antioxidant and antifungal properties (El-Shiekh et al., 2024). Oleanolic acid exhibits pharmacological properties including antidiabetic, anti-inflammatory, antioxidant, immune-boosting, hepatoprotective, anticancer, and antiviral effects (Günther & Bednarczyk-Cwynar, 2025; Wasim & Bergonzi, 2024). It possesses significant antioxidant properties that can be used to prevent many diseases related to oxidative stress (Günther & Bednarczyk-Cwynar, 2025). Widdrol has shown potent anticancer effects in human colon adenocarcinoma cells in vitro by inhibiting cell growth and inducing apoptosis (Kwon et al., 2010). Similarly, another in vitro study demonstrated its anti-cancer activity in colon cancer cells by inducing cell death (Kim, 2012). α-Pinene, one of the isomers of Pinene, is a vital monoterpenoid with anticancer activities against human ovarian cancer cell lines and hepatocellular carcinoma cell lines. It can induce apoptosis, disrupt ROS formation, and display several inhibitory effects on hepatocellular carcinoma cell lines, including BEL-7402 cells and HepG2 cells under in vitro conditions (W. Chen et al., 2015; J.-B. Yang et al., 2016). It also exerts anti-inflammatory and antioxidant properties. Meanwhile, the other isomer, β-Pinene, is also observed to have inhibitory effects on leukemia, breast cancer, and non-small cell lung cancer (combined with paclitaxel) (Salehi et al., 2019). Linalool has been shown to possess potent anticancer activity in an ovarian cancer model and in hepatocellular carcinoma cell lines (Kłos & Chlubek, 2022). It also showed anticancer potential in leukemia and melanoma (Ansari & Akhtar, 2019). Other sesquiterpenoid compounds, such as β-caryophyllene and cis-Nerolidol, possess various biological effects, including anti-inflammatory, antioxidant, and antimicrobial properties (Chan et al., 2016; Gushiken et al., 2022), while 6 Epi beta bisabolol has anti-inflammatory activity (Egbuta et al., 2022). As these compounds mainly possess antioxidant and chemopreventive properties, they may effectively neutralize ROS and induce cell death. Due to their lipophilic and hydrophobic nature, these compounds are capable of interacting with the lipid bilayer and can target multiple intracellular signaling pathways. Additionally, they can modulate the MAPK signaling pathway, which plays a critical role in the activation and progression of the oral carcinogenetic cascade (Kumar & Jha, 2023; Sharmila et al., 2025). Through these complementary mechanisms, the compounds may serve as supportive agents in the management of OSCC.
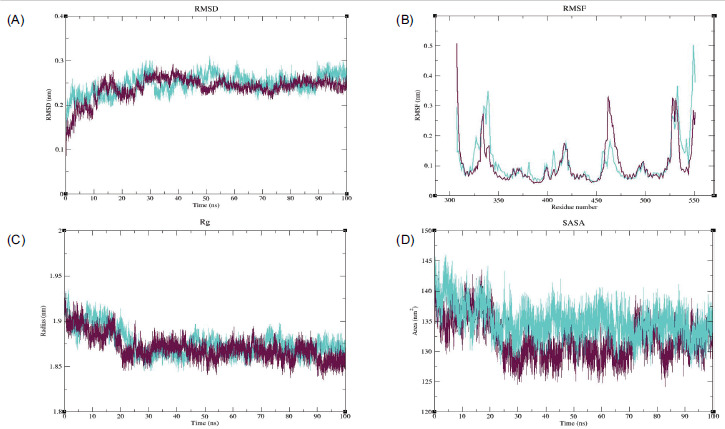
Molecular docking of the OSCC targets with their respective phytochemicals revealed strong binding interactions. The highest binding energies were observed for the MMP9–Geranyl butyrate, PTGS2–Ledol, and ESR1–(-)-Globulol complexes. Geranyl butyrate and Ledol were also found to interact with MMP2 and ESR1. Similarly, ESR1 was targeted by multiple compounds, including Widdrol, Oleanolic acid, Linalool, alpha-Pinene, beta-Pinene, beta-Caryophyllene, and 6-epi-beta-bisabolol. All these compounds exhibited strong binding interactions with the OSCC targets (Table 1). Metalloproteinases (MMP2 and MMP9), which play a major role in degrading ECM to facilitate tumor metastasis in OSCC, were effectively targeted by the compounds of S. cumini. MD simulations were performed for the top three protein–ligand complexes to assess their binding stability. The MMP9–Geranyl butyrate complex exhibited stable and low RMSD values, indicating greater stability. RMSF analysis revealed less flexibility across major residues, while Rg confirmed the compactness of the complex. SASA analysis also confirmed that the ligand-bound complex had a more stable conformation with less solvent exposure (Figure 6). The PTGS2–Ledol complex exhibited lower RMSD values, indicating a more stable form than the Apo structure. RMSF analysis showed that the complex had less flexibility compared to its Apo form. Additionally, Rg and SASA analyses revealed that the complex attained a more compact and stable conformation, as evidenced by its lower Rg and SASA values (Figure 7). In the ESR1–Globulol complex, RMSD, RMSF, Rg, and SASA analyses revealed that both the complex and the Apo form exhibited similar fluctuations throughout the simulation period (Figure 8).
The compounds Geranyl butyrate, Ledol, and (-)-Globulol contributed to the stabilization of their respective protein targets. The bioactive compounds of S. cumini interacted with multiple targets that are clinically associated with tumorigenesis in OSCC, indicating their potential translational relevance. The network pharmacology approach enabled the identification of multi-target effects, which is a key advantage of computational approaches. Nevertheless, this study has certain limitations. It is solely based on in silico approaches and is therefore considered hypothesis-generating. Important parameters such as release profile index (RPI), long-term toxicity, enhanced permeability and retention (EPR) effects, and pharmacokinetic behavior, which are critical for translational assessment, were not evaluated in this study and will be investigated in future research (Bommanavar et al., 2025). Further experimental studies using oral cancer cell lines and in vivo models are needed to validate these findings and assess their biological and clinical relevance. Overall, this network pharmacology-based approach improves our understanding of the mechanisms of S. cumini compounds and highlights their potential therapeutic role in OSCC.
Conclusion
In this study, a network pharmacology approach was employed to explore the pharmacological and molecular mechanisms of Syzygium cumini for treating OSCC. Through a Venn diagram, 290 common targets associated with OSCC and S. cumini were identified. Several important biological processes, such as hormone regulation, inflammatory response, and MAPK signaling, were identified through enrichment analysis, which play a critical role in OSCC. The core targets identified from the network have significant relevance and importance in the context of OSCC. The six targets (MAPK1, PTGS2, ESR1, MMP2, MMP9, and PARP1) identified from the gene–disease association analysis were considered the top targets, and their corresponding bioactive compounds were docked against them. Geranyl butyrate, Ledol, and (-)-Globulol were identified as the key bioactive compounds that exhibited strong binding affinity with the targets. Furthermore, MD simulations confirmed the stable binding interactions of these compounds with the OSCC targets, suggesting their potential roles as therapeutic agents for OSCC. However, further validation is required to support these computational findings.