
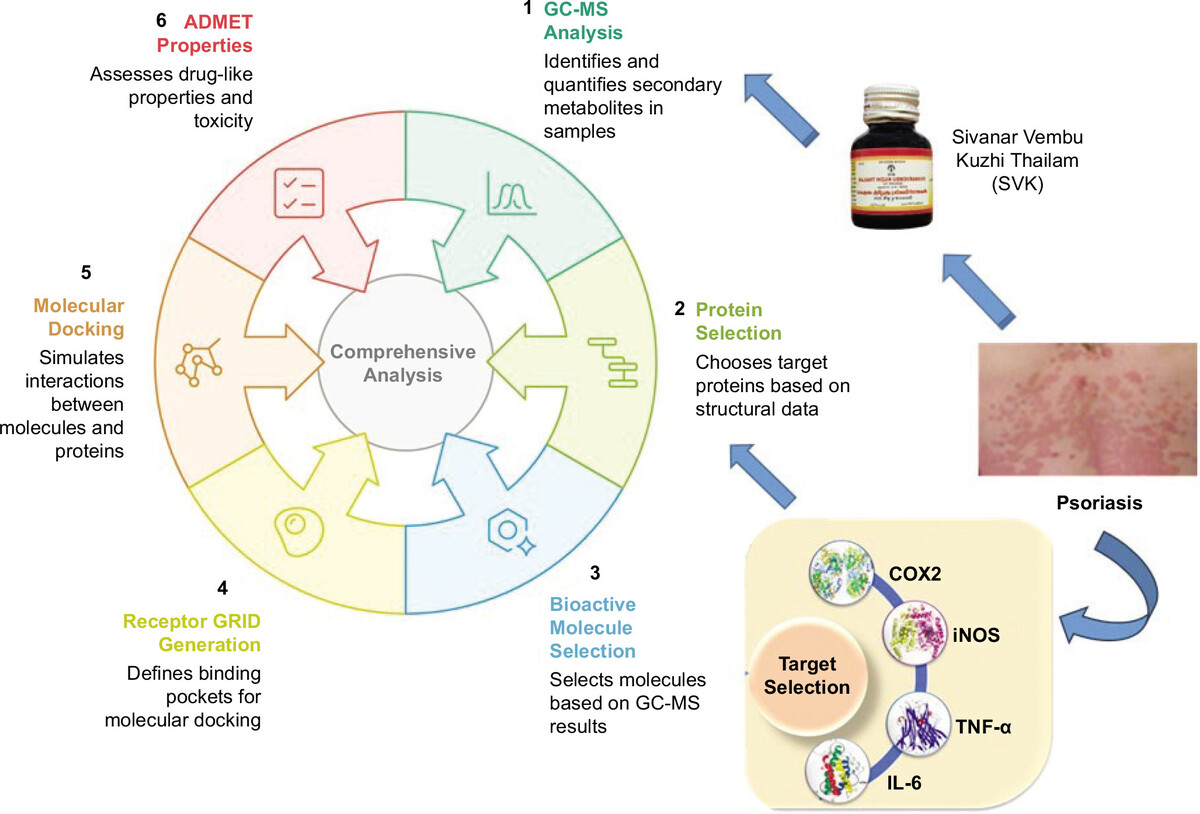

1. Introduction
Psoriasis is a chronic inflammatory skin disorder that arises primarily due to a combination of hereditary susceptibility and ageing factors. It is characterized by sharply demarcated plaques with white, scaly, indurated, pruritic, and often painful skin plaques (Kamiya et al., 2019). Psoriasis predominantly affects adults and exhibits an uneven global distribution. Its prevalence is higher in high-income countries and regions with ageing populations. Variability in prevalence is influenced by multiple factors, including age, sex, geographic location, ethnicity, genetic predisposition, and environmental determinants (Parisi et al., 2020). Clinically, cutaneous psoriasis can be classified into psoriasis vulgaris, inverse psoriasis, guttate psoriasis, pustular psoriasis, palmoplantar pustulosis, and erythrodermic psoriasis (Yan et al., 2021). Other systemic conditions such as obesity, hypertension, hyperlipidemia, diabetes, metabolic syndrome, cardiovascular diseases, and chronic renal diseases frequently coexist with psoriasis (Tokuyama and Mabuchi, 2020).
The hallmark of psoriasis is sustained inflammation that leads to uncontrolled keratinocyte proliferation and dysfunctional differentiation. The pathogenesis of psoriasis involves antimicrobial peptides (AMPs), dendritic cells (DCs), tumour necrosis factor-alpha (TNF-α), interleukin (IL) 23, IL17, IL22, Th17, and signal transducer and activator of transcription (STAT) 3 (Nestle et al., 2005). Genetic factors play a significant role in the pathogenesis of psoriasis, with a strong familial component observed in many cases, indicating that certain genes, viz. COX 2, IL-6, iNOS, and TNF-α, make individuals more susceptible to developing the condition (Conrad and Gilliet, 2018). T-cells, a type of white blood cell, become overactive and trigger inflammation in the skin. These cells release cytokines such as TNF-α, IL-17, and IL-23, which contribute to the inflammatory process. The cytokines stimulate the rapid proliferation of keratinocytes, leading to the formation of thick and scaly plaques (Nestle et al., 2005).
AMPs are short proteins that protect against pathogens and modulate inflammation (Morizane and Gallo, 2012). In psoriasis, AMPs like β-defensins, S100 proteins, and cathelicidin (LL37) are overexpressed, impacting immune response. The skin hosts a variety of DCs, such as Langerhans cells (LCs), dermal conventional DCs (cDCs), plasmacytoid DCs (pDCs), and inflammatory DCs (iDCs), all of which are essential for healthy skin and immune function. LCs present antigens, cDCs activate T cells, pDCs produce interferon-alpha (INFα) for antiviral defense, and iDCs secrete cytokines like TNFα, IL-12, and IL-23, which contribute to psoriasis pathogenesis (Chen et al., 2016). Lifestyle factors such as obesity, smoking, and excessive alcohol consumption can increase the severity and frequency of psoriasis outbreaks. Hormonal level changes, such as puberty or menopause, can influence psoriasis (Michalski et al., 2023). Certain medications also play a vital role in the onset or exacerbation of symptoms, highlighting the complex interaction between external factors and the immune system in disease pathogenesis.
The Siddha system is one of the ancient traditional Indian medicinal practices and is distinctively rooted in the Southern region of India. Sivanarvembu Kuzhi Thailam (SVK) is one of the classical Siddha formulations used for various skin diseases. The ingredients are Sivanaarvembu (SV) (Indigofera asplathoides), Akasa Karudan Kizhangu (Corallocarpus epigaeus), Valuluvai arisi (Celastrus paniculatus), and Karpooram (Cinnamomum amphora) (Siddha Formulary of India, 1992). A special technique called the Kuzhi thailam method (a traditional technique of destructive distillation) is used to extract the oil. The phytochemical analysis of SVK reveals the presence of steroids, triterpenes, alkaloids, phenolic groups, tannin, glucose, catechins, proteins, and glucose (Ahmed et al. 2019). Ahmed Sundar (2017) conducted preliminary research on Sivanarvembu chooranam and concluded it to be an anti-inflammatory, analgesic, and anti-histaminic drug for psoriatic patients (Ahmed, 2017). Although Siddha medications are widely used for psoriasis, there are no published studies on their safety and therapeutic effectiveness (Amuthan, 2020). This study bridges traditional Siddha knowledge with modern scientific techniques by integrating phytochemical analysis, GC-MS profiling, and in silico molecular docking to elucidate the therapeutic potential and anti-inflammatory mechanism of SVK in psoriasis management.
The intact structural and biochemical diversity in natural compounds has rendered them indispensable in psoriatic therapy, initially due to their minimal side effects. The study aimed to perform Gas Chromatography–Mass Spectrometry (GC-MS) fingerprinting to analyse the biomolecules in SVK, providing a detailed chemical profile. In silico molecular docking was conducted to examine the binding of SVK metabolites with inflammatory proteins COX 2, iNOS, IL-6, and TNF-α, which play a crucial role in psoriasis. Using the Schrödinger software suite, the interaction between SVK compounds and these proteins was evaluated, offering insights into SVK’s potential anti-inflammatory mechanism against psoriasis.
2. Materials and methods
2.1. Instrumentation parameters of the GC-MS analysis
Gas Chromatography–Mass Spectrometry (GC-MS) analysis was performed to identify the secondary metabolites present in the SVK. The Agilent 8890 GC was inert, non-locked, with dimensions of 30 m length, 250 µm diameter, and 0.25 µm film thickness. Helium gas was utilized as the carrier gas at a constant flow rate of 1.2 mL/min to separate sample components. The maximum oven temperature was maintained at 350°C throughout the chromatographic run. A 1 µL sample volume was injected into the column injector reservoir. The oven temperature program started at 180°C, held for 3 min, and ramped up at 5°C/min to 300°C, where it was held for 5 min. Detection was performed using the Agilent 5977 GC/MS module. The mass spectrum provides information about the molecular structure of the compounds. The quadrupole temperature was set at 150°C, while the ion source temperature was 230°C. Ionization was achieved in electron impact mode at 70 eV. Mass spectrometry data were acquired in scanning mode from 50 to 600 amu, with a 150 threshold and a scan speed of 1,562 [N=2]. Identified components were compared against the National Institute of Standards and Technology (NIST) GC-MS library for known spectra (Gunalan et al., 2020).
2.2. Identification of compounds
GC-MS was used to identify the compounds by separating them in gas chromatography based on their chemical properties and then analysing the resulting mass spectra to determine their molecular structure and identity. We determined the relative abundance of each component by calculating the percentage based on comparing the average peak area of each component to the total peak area obtained from the chromatogram. This method enabled us to quantify the contribution of each compound within the mixture, providing insights into its composition and distribution.
2.3. Selection and preparation of target proteins and bioactive molecules for molecular docking studies
In this study, four target proteins, COX 2, iNOS, TNF-α, and IL-6, were selected for computational analysis. These proteins were obtained from the RCSB Protein Data Bank database (RCSB PDB) (https://www.rcsb.org/) with the following PDB IDs: 5IKR for COX 2, 3E7G for iNOS, 2AZ5 for TNF-α, and 1ALU for IL-6. The selection was based on the proteins’ high-resolution structures, ensuring the availability of high-quality structural data for docking analysis. The obtained target protein structures were imported into the Maestro interface to ensure structural correctness using the Protein Preparation Wizard within the Schrödinger suite, employing default settings. During this process, heteroatoms, crystallized water molecules, and co-crystallized ligands were removed. Hydrogen atoms were added, and any missing side chains were filled using the Prime module. The three-dimensional (3D) structures of the selected proteins were then stabilized using the OPLS3 force field, ensuring optimal geometry and energy minimization for accurate computational analysis.
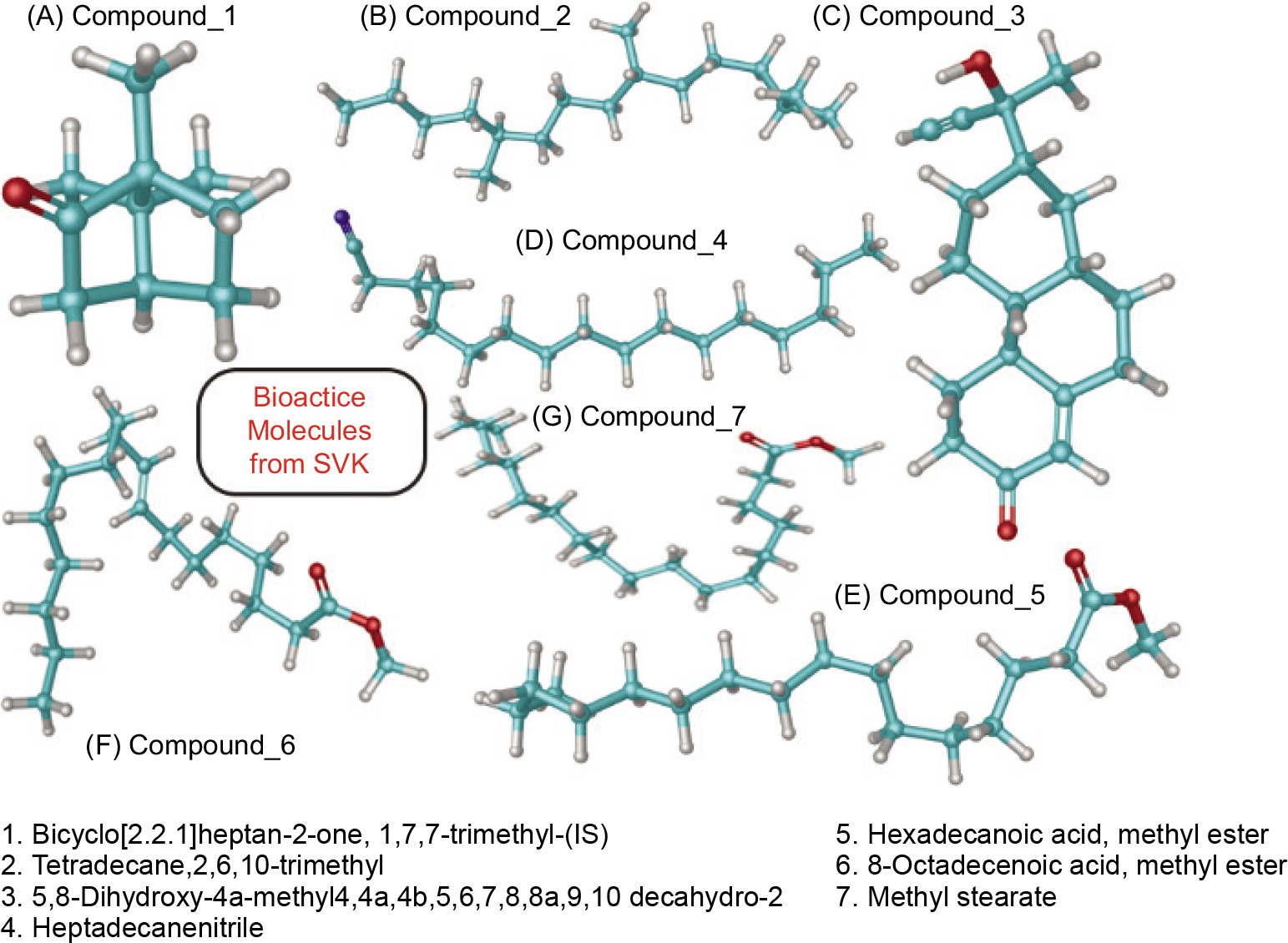
Bioactive molecules were selected based on their area percentage in the GC-MS chromatogram. Area percentages >5% were considered for the docking studies (depicted in Figure 1). These molecules were cross-validated using the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) to ensure their authenticity and to retrieve their three-dimensional (3D) coordinates in SDF format. The obtained 3D coordinates of the ligand molecules were then imported into the LigPrep module of the Schrödinger suite for further preparation. LigPrep facilitated the correction and optimization of the ligand structures, addressing issues such as improper valency, missing hydrogen atoms, and incorrect bond orders. This initial correction step ensured that all ligands were chemically accurate and ready for detailed computational studies. Subsequently, hydrogen atoms were added to the structures to ensure proper protonation states and overall charge balance of the molecules. This step is critical for accurately simulating physiological conditions during molecular docking (Jayaraman et al., 2024). To further enhance the reliability of the docking studies, possible tautomeric forms of the ligand molecules were generated using the Epik tool within the Schrödinger suite. Epik predicts and generates all relevant tautomers, ensuring that the most biologically active forms of the ligands are considered in the docking simulations.
2.4. Receptor GRID generation
Before molecular docking, it was crucial to accurately identify the binding pockets or active site residues of the target proteins. This step ensures that the GRID box properly encompasses the binding site, allowing ligand molecules to interact effectively with the catalytic site of the protein. Information regarding the active site residues was collected from the CastP server (http://cast.engr.uic.edu) and through the analysis of co-crystallized ligand molecules. The CastP server provides detailed insights into the topography and volume of protein cavities, which is essential for defining the precise location and dimensions of the active sites. Utilizing both computational data from CastP and empirical data from co-crystallized ligands for GRID generation ensures a comprehensive understanding of the active sites, thereby enhancing the accuracy and reliability of the molecular docking studies.
2.5. Molecular docking
In this study, the molecular docking protocol was executed using the GLIDE extra precision (XP) docking module, a sophisticated tool within the Schrödinger suite. GLIDE XP offers enhanced accuracy in docking simulations by employing a comprehensive scoring function that considers various interaction parameters, such as hydrogen bonding, hydrophobic interactions, and electrostatic forces. This approach ensures a more precise prediction of ligand binding affinities and orientations, thus providing reliable insights into the molecular interactions at the active sites of the selected target proteins (Owoloye et al., 2022). To further analyze the docked complexes, both 3D and two-dimensional (2D) visualizations of intermolecular interactions were employed. The UCSF Chimera tool (www.rbvi.ucsf.edu/chimera) was used to visualize the 3D interactions, offering a detailed view of conformational changes within the protein–ligand complexes. Additionally, the Schrödinger suite generated 2D interaction diagrams, which were inspected to understand the specific binding interactions, such as hydrogen bonds, hydrophobic contacts, and electrostatic interactions, between the ligands and the protein residues. This comprehensive visualization and analysis approach ensures a thorough understanding of the docking results, aiding in the refinement and optimization of potential therapeutic compounds.
2.6. Pharmacokinetics profile assessment—ADMET properties
In drug design and development, assessing the pharmacokinetics profile, particularly the ADMET properties (Absorption, Distribution, Metabolism, Excretion, and Toxicity) of candidate molecules, is an essential step before proceeding to clinical trials. These properties provide critical insights into the behaviour of the drug within the human body and its overall safety and efficacy (Jayaraman et al., 2021). In this study, the QikProp module within the Schrödinger suite was utilized to predict the pharmacokinetic properties of the candidate molecules. QikProp is known for its robust and reliable predictions, offering valuable data on a wide range of ADMET parameters. Additionally, biophysical characteristics such as synthetic accessibility, molecular refractivity, and bioavailability score were predicted using the free online tool Swiss ADME (http://www.swissadme.ch/). Swiss ADME provides a comprehensive evaluation of drug-likeness and other relevant pharmacokinetic properties, aiding in the selection and optimization of potential drug candidates. The toxicity profile, a crucial aspect of the pharmacokinetic assessment, was evaluated using the ProTox II web server (https://tox.charite.de/protox3/). By submitting the SMILES (Simplified Molecular Input Line Entry System) notation of each ligand molecule, ProTox II predicts various toxicity endpoints, including organ toxicity, toxicological pathways, and potential side effects. This thorough toxicity assessment ensures that only the safest and most promising candidates move forward in the drug development process. By integrating these advanced computational tools, this study ensures a comprehensive and accurate assessment of the pharmacokinetics and toxicity profiles of the candidate molecules, thereby enhancing the likelihood of success in subsequent clinical trials.
3. Results
3.1. GC-MS fingerprinting
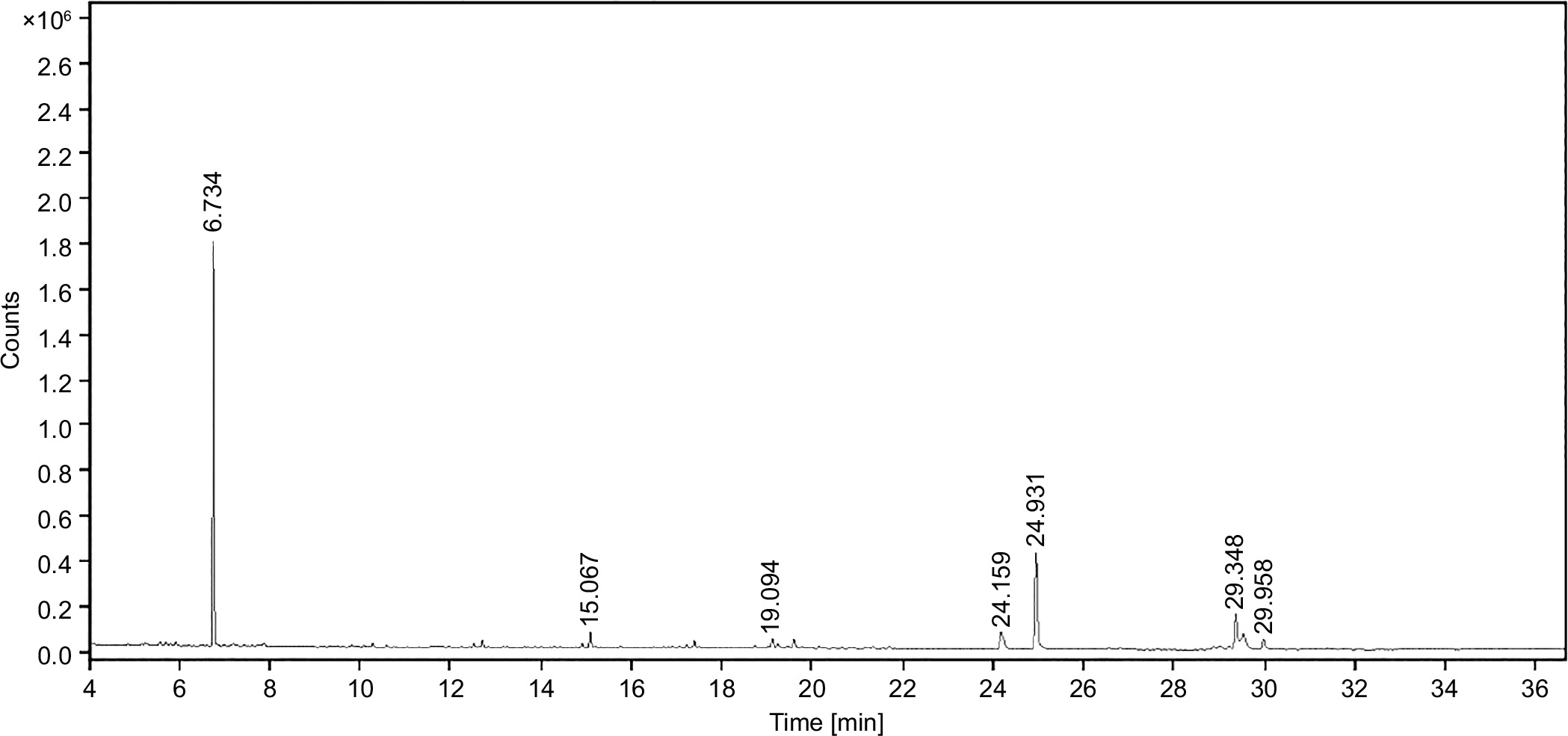
GC-MS can identify and quantify the individual compounds present in the sample of interest, providing a detailed chemical profile. The purity of any drug can also be assessed by detecting any impurities present in the drug molecules. It helps to ensure the quality of the drug by comparing the chemical compounds across different batches. It plays a major role in identifying the bioactive compounds that are responsible for the therapeutic effects of the drug. Additionally, GC-MS aids in the discovery and development of novel drugs by providing insight into the chemical constituents. GC-MS fingerprinting analysis of the SVK has displayed seven distinguishable peaks (Figure 1). The peaks observed in the chromatogram were meticulously compared against mass spectra available in the NIST 2017 library. A detailed summary of the identified phytoconstituents was depicted in Table 1, encompassing essential details such as their chemical names, retention time (RT), molecular formulas, molecular weights, and the relative peak areas expressed as percentages.
Table 1
List of the components present in the SVK.
All seven secondary metabolites exhibited distinct RTs and varied area percentages. From this study, metabolites with area percentage exceeding 5% were deemed significant. The major component present in the SVK was Bicyclo [2.2.1] heptan-2-one, 1,7,7-trimethyl-(IS) (compound 1), which had the highest area percentage of 57.27%. This was followed by hexadecanoic acid, methyl ester (compound 5) (21.98%), 8-octadecenoic acid, methyl ester (compound 6) (8.07%), and heptadecane nitrile (compound 4) (6.22%). The remaining three metabolites—Tetradecane, 2,6,10-trimethyl (compound 2); Methyl stearate (compound 7); and 5,8-Dihydroxy-4a-methyl-4,4a,4b,5,6,7,8,8a,9,10 decahydro-2 (compound 3)—had area percentage of 2.49%, 2.25%, and 1.72%, respectively, and hence were considered non-significant due to the presence of a negligible amount.
3.2. Molecular docking analysis
Molecular docking analysis is a critical technique for elucidating how potential drug molecules interact with their biological targets, which is essential for identifying and optimizing promising therapeutic candidates. In this study, molecular docking was performed using the GLIDE XP module of the Schrödinger suite, known for its high accuracy in predicting ligand–receptor interactions. Initially, bioactive molecules (Figure 2) were selected based on the area percentage observed in the GC-MS chromatogram analysis. The selected targets included COX 2 (PDB ID: 5IKR), iNOS (PDB ID: 3E7G), TNF-α (PDB ID: 2AZ5), and IL-6 (PDB ID: 1ALU). These proteins were chosen due to their significant roles in inflammatory and immune responses, making them relevant targets for therapeutic intervention for psoriasis. The docking results were presented in Table 3 for COX 2. Compound 3 showed the highest binding affinity with a docking score of –7.732 kcal/mol, followed by compound 2 with a score of –5.116 kcal/mol. The iNOS docking analysis revealed the highest binding energy of –6.50 kcal/mol, indicating a strong interaction between the ligand and SVK. For IL-6, the docking scores ranged from 0.31 to –3.229 kcal/mol (Table 3), suggesting comparatively weaker interactions than those observed for iNOS and COX 2. The TNF-α docking scores ranged from 0.95 to -3.486 kcal/mol, indicating a relatively lower level of bonding between the ligands and the target protein.
3.3. Molecular interaction analysis
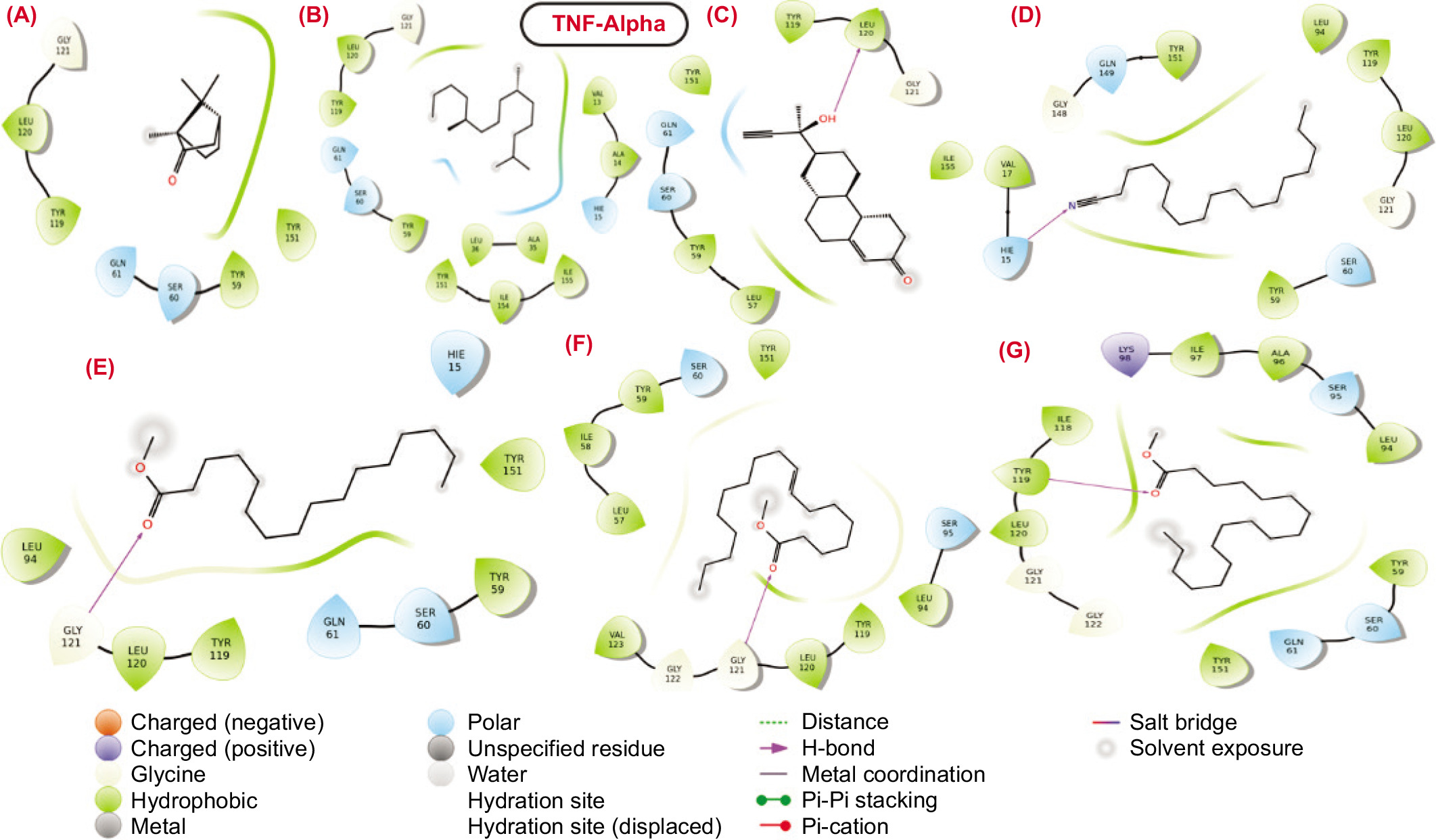
The molecular interactions between the binding pocket residues of the selected proteins and the bioactive molecules are illustrated in Figures 3–6. The analysis of the molecular interaction profiles suggests that binding was established through conventional hydrogen bonds, hydrophobic interactions, salt-bridge contacts, stacking interactions, and other types of non-covalent interactions.
Figure 3
2D structural representation of molecular interaction between the bioactive molecules from SVK against COX-2 protein.
Figure 4
2D structural representation of molecular interaction between the bioactive molecules from SVK against IL6 protein.
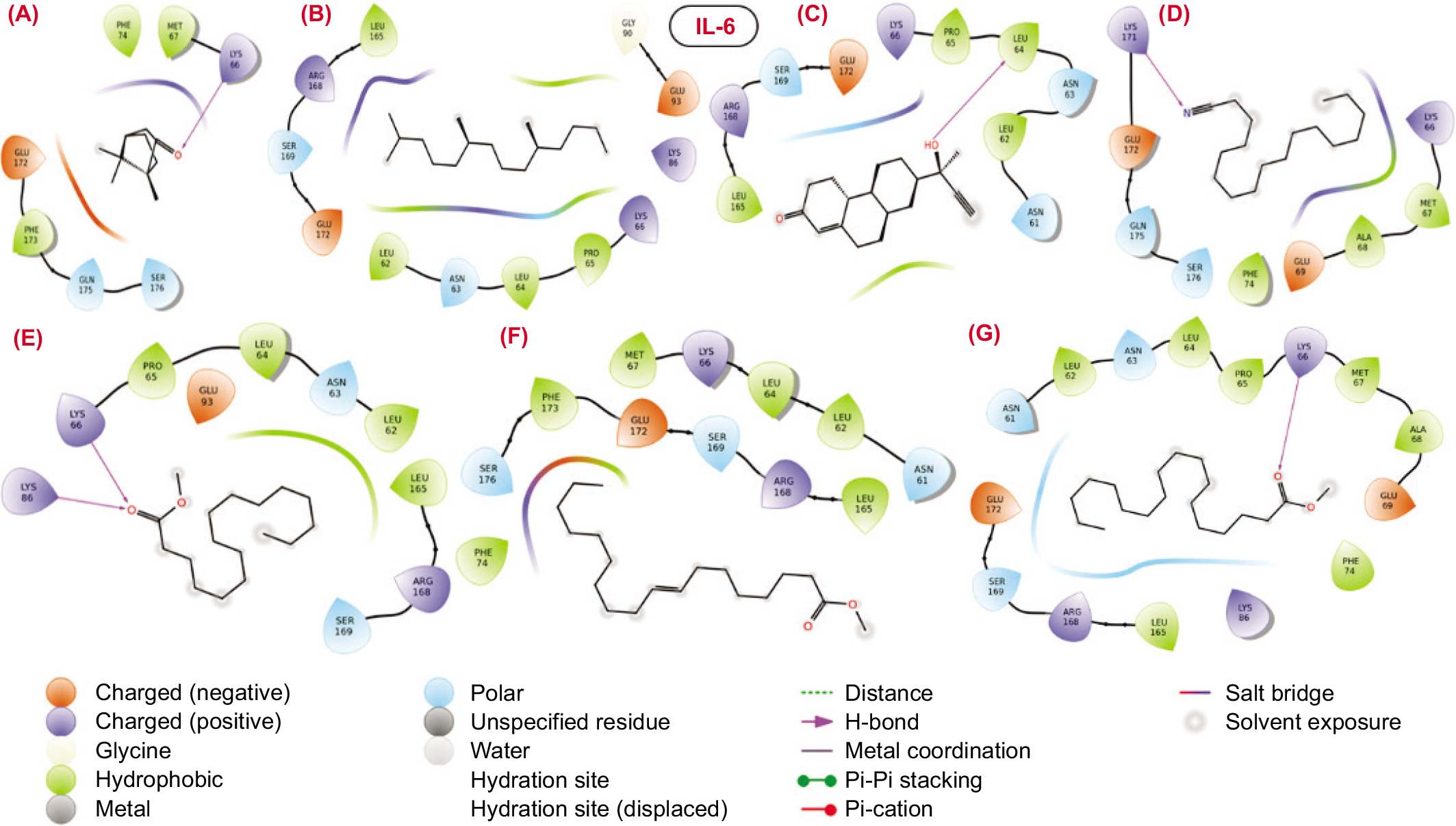
Figure 5
2D structural representation of molecular interaction between the bioactive molecules from SVK against iNOS protein.
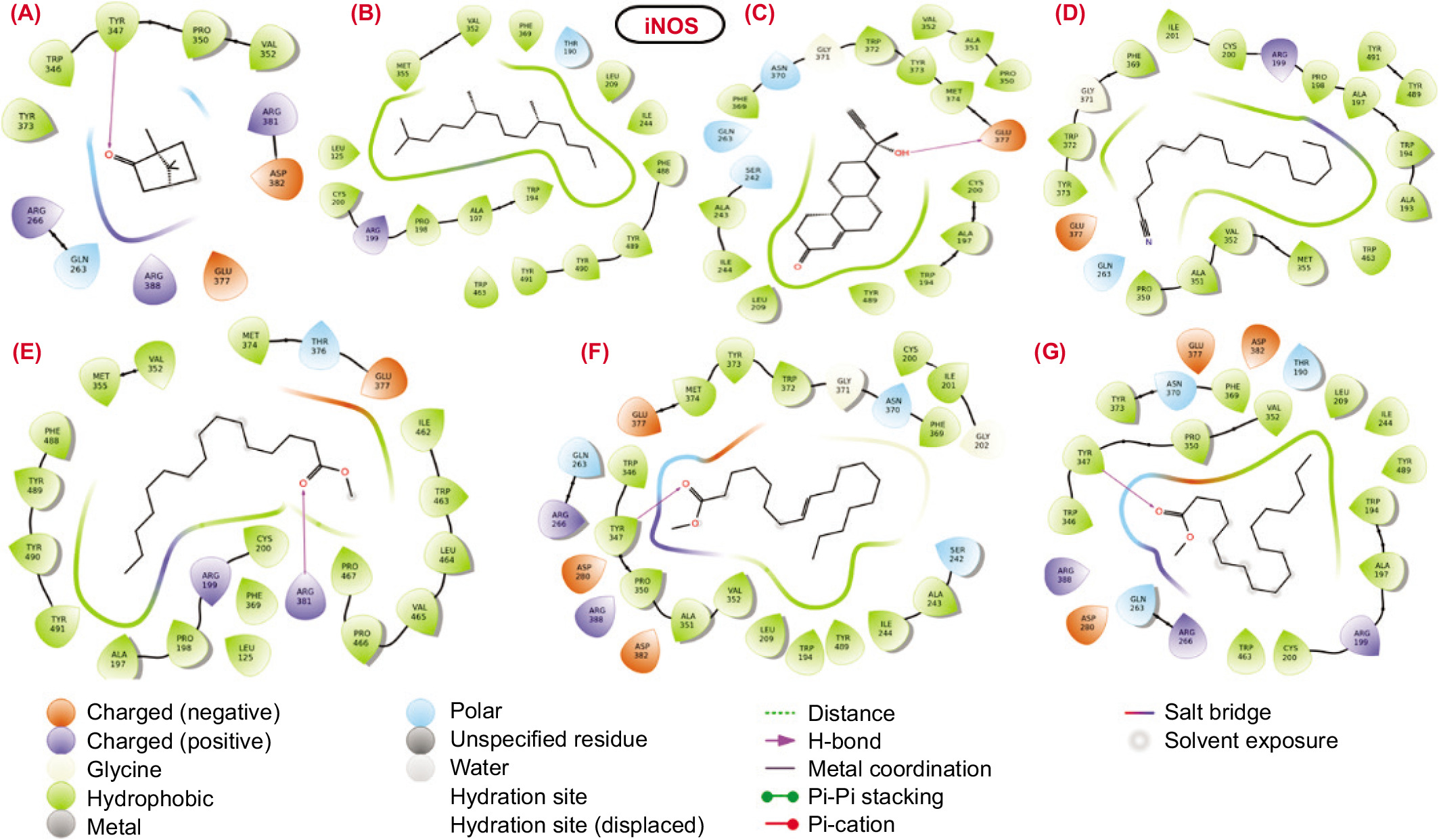
Figure 6
2D structural representation of molecular interaction between the bioactive molecules from SVK against TNF-Alpha protein.
COX 2: Unlike the other proteins, the interacting complexes of bioactive molecules with the COX 2 binding site residues showed that only compound 1 and compound 3 were involved in hydrogen bond (H-bond) interactions, with docking scores of 4.081 and –7.732, respectively, as shown in Table 2. None of the remaining molecules demonstrated significant H-bond contacts with the COX 2 protein. In the complexes formed by compound 1 and compound 3, the positively charged residue Arg120 actively engaged in H-bond interactions with the carbonyl groups (C=O) of these compounds. Additionally, a significant number of amino acid residues were involved in hydrophobic contacts, contributing to the structural stability of these bioactive molecules.
Table 2
Molecular interaction profile and docking score of bioactive molecules from SVK against selected protein target for COX 2.
IL6: The docked complexes of bioactive molecules with IL-6 protein revealed that hydrogen bonds and hydrophobic interactions are the dominant forces contributing to the stability of the complexes. Specifically, compounds 1, 5, and 7 established hydrogen bonds with the positively charged residue Lys66. Additionally, compound 5 also formed a hydrogen bond with Lys86. Compounds 3 and 4 formed hydrogen bonds with the hydrophobic residue Leu64 and the positively charged residue Lys171. Compound 3 showed a docking score of approximately -3.229, as presented in Table 3. However, compounds 2 and 6 did not participate in hydrogen bond interactions with the binding pocket residues of IL-6. Instead, these compounds primarily engaged in hydrophobic contacts.
Table 3
Molecular interaction profile and docking score of bioactive molecules from SVK against selected protein target for IL6.
iNOS: The molecular interaction analysis of the docked complexes between the iNOS protein and the bioactive molecules revealed that all the compounds formed a single hydrogen bond with the binding pocket residues of iNOS, except for compounds 2 and 4. Notably, three compounds—compound 1, compound 6, and compound 7—engaged in hydrogen bond interactions with the hydrophobic residue Tyr347, underscoring its significance in maintaining the structural stability of the complexes. Meanwhile, compound 5 and compound 3 established hydrogen bond contacts with the positively charged Arg381 and the negatively charged Glu377, with the docking scores of -2.26 and -6.50, respectively.
Despite showing considerable docking scores, compound 2 (-4.269) and compound 4 (-2.26) did not exhibit any hydrogen bonds in their interaction networks (Table 4). Instead, these compounds demonstrated numerous hydrophobic interactions, highlighting the critical role of hydrophobic contacts in the structural stability of the complexes. This emphasizes the importance of hydrophobic interactions, alongside hydrogen bonds, in stabilizing the bioactive molecule–iNOS protein complexes.
Table 4
Molecular interaction profile and docking score of bioactive molecules from SVK against selected protein target for iNOS.
TNF-α: Although the molecular docking scores for the bioactive molecules against the TNF-α protein were generally unfavourable and low, all molecules except for compound 1 and compound 2 exhibited hydrogen bond interactions with the binding pocket residues of the target protein. Specifically, compound 3 and compound 7 engaged in hydrogen bond interactions with the hydrophobic residues Leu120 and Tyr119, respectively. Additionally, the polar residue HIE15 of TNF-α formed a hydrogen bond with the nitrile group (C≡N) of compound 4. The docking scores for compound 1 and compound 3 were -2.311 and -3.486, respectively, as shown in Table 5. Furthermore, the carbonyl groups (C=O) of compound 5 and compound 6 actively participated in hydrogen bond interactions with the residue Gly121. This analysis highlights that, despite the low docking scores, several bioactive molecules still exhibit significant hydrogen bond interactions with key residues in the TNF-α binding pocket, suggesting potential avenues for improving binding affinity and stability through targeted structural modifications.
Table 5
Molecular interaction profile and docking score of bioactive molecules from SVK against selected protein target for TNF-α
3.4. ADME-T properties prediction
Ensuring the clinical safety profile of new therapeutic compounds is paramount for the successful progression of drug discovery and development. The molecular weight of all bioactive molecules from SVK falls below 500 Da, indicating a promising pharmacokinetic profile and aligning with established guidelines for drug-likeness. Additionally, the H-bond donor capabilities of all bioactive molecules are within the optimal range of 0–6. However, compounds 1, 2, and 4 did not meet the H-bond acceptor criteria, which require values within the range of 2–20 (Table 6). Interestingly, the human oral absorption (HOA) values for all bioactive molecules were classified as high (class 3) with an HOA percentage of 100%, except for compound 7, which showed an HOA percentage of 91.26%. This high HOA classification indicates a strong potential for oral bioavailability for most of the analyzed compounds, suggesting their promising efficacy as orally administered drugs. Furthermore, all bioactive molecules from SVK exhibited high MDCK and CaCo-2 values, exceeding 500. MDCK cells are considered an excellent mimic for the blood–brain barrier, while CaCo-2 cells serve as a reliable model for the intestinal barrier.
Table 6
Predicted pharmacokinetics of SVK oil ligands using Swiss ADME tool.
The biophysical properties of SVK were effectively predicted using the SwissADME computational tool, and the results are presented in Table 7. According to the analysis, compound 2 exhibited a low gastrointestinal (GI) absorption rate compared to the other molecules, which showed high GI absorption values. The compounds were also assessed for P-glycoprotein (P-gp) substrate activity, where a response of “Yes” indicates that the compound is likely to be transported by P-gp, potentially affecting its absorption and distribution. Cytochrome P450 (CYP) enzymes, a critical family involved in drug detoxification and xenobiotic metabolism in the liver, were evaluated for interactions with SVK ligands. The results indicated that the SVK ligands did not inhibit CYP2C19, CYP2D6, or CYP3A4. However, compounds 4 to 7 selectively inhibited CYP1A2, while compounds 2 and 3 inhibited CYP2C9. These findings, summarized in Table 7, highlight the variability in biophysical properties among the SVK ligands, their potential effects on drug metabolism, and their interactions with key metabolic enzymes.
Table 7
Pharmacokinetic properties prediction for the bioactive molecules obtained from the SVK through QikProp module of Schrödinger suite.
The detailed toxicity profiling of bioactive molecules is presented in Table 8. This thorough assessment helps to identify any potential risks associated with the therapeutic agents and represents a vital step in the drug development process. Notably, four bioactive molecules—compound 3, compound 5, compound 6, and compound 7—were classified as Class V, indicating a low likelihood of causing toxicity if ingested. In contrast, compounds 2 and 4 were classified as Class III, and compound 1 falls into Class IV, suggesting a higher potential for harm if swallowed. The toxicity profile analysis also suggested that all of the bioactive molecules exhibited no hepatotoxicity, cytotoxicity, carcinogenicity, mutagenicity, or immunotoxicity. This comprehensive assessment underscores the potential safety of these compounds and supports their further development as therapeutic agents.
Table 8
Toxicity properties of bioactive molecules obtained from SVK through ProTox-II server.
4. DISCUSSION
GC-MS can be used to study the metabolites of traditional medicines in the body, providing insights into how these medicines are absorbed, distributed, metabolized, and excreted. In this study, we investigated the bioactive compounds and anti-inflammatory properties present in SVK through GC-MS analysis, and several compounds were identified. Based on area percentage, seven compounds were identified as major ones. Among these seven compounds, the most abundant was compound 1, commonly known as Bicyclo [2.2.1] heptan-2-one, 1,7,7-trimethyl-(IS), referred to as Camphor. Camphor is widely recognized for its use as an embalming agent and as an active ingredient in antiseptic creams due to its antimicrobial properties. It is readily absorbed into the skin and also acts as a local anaesthetic agent (Shabir and Bradshaw, 2010). The second major compound, hexadecanoic acid, methyl ester, is commonly known as palmitic acid methyl ester (PAME) (Ahunna, 2015). PAME has antibacterial properties (Shaaban et al., 2021). Kalpana et al., (2012) have reported that n-hexadecanoic acid has antioxidant, anti-inflammatory, hypocholesterolemic, and cancer-prevention activities. 8-octadecenoic acid, methyl ester (compound 6) (8.07%) and heptadecane nitrile (compound 4) (6.22%) were the other major compounds in SVK. Various researchers have demonstrated diverse biological activities such as anti-inflammatory, antibacterial, hypocholesterolemic, hepatoprotective, antioxidant, and antimicrobial properties (Rahman et al., 2014). The synergy among multiple metabolites may enhance the overall bioactivity of SVK, contributing to their therapeutic effects towards psoriasis.
Molecular docking is a pivotal computational technique in drug design, employed to predict the optimal binding orientation of ligand molecules with their receptor proteins and to elucidate the interaction mechanisms between ligands and targeted proteins (Gunalan et al., 2014). This method is crucial for understanding how potential drug molecules interact with their biological targets, thereby facilitating the identification and optimization of promising therapeutic candidates. This study utilized docking experiments to identify the topmost lead molecule that can modulate a protein’s activity by either activating or inhibiting it. These potential modulators exhibit strong binding affinities and structural compatibility, indicating their effectiveness. Further investigation into these compounds will help to clarify their mechanisms of action, potentially leading to new therapeutic developments (Agu et al., 2023).
The docking scores calculated by GLIDE XP for the bioactive molecules against each target protein are presented in Tables 2–5, demonstrating a wide range from 0.31 to –7.73 kcal/mol. These scores indicate the binding affinity and potential effectiveness of the ligands in interacting with their respective targets. Cyclooxygenase-2 (COX 2) is an inducible cyclooxygenase, and overexpression of COX 2 can promote cell proliferation and angiogenesis (Greenhouse et al., 2009). Immunohistochemistry also demonstrates that the expression of COX-2 is upregulated in psoriatic lesions (Yalcin et al., 2007). Wang et al., (2016) demonstrated that the mRNA and protein levels of COX 2 in psoriatic skin were higher than those of the normal control group, and this confirms that COX 2 is likely involved in the development of psoriasis. Inducible NO synthase (iNOS) will be expressed only after cell activation through various inflammatory mediators like cytokines, lipopolysaccharide (LPS), etc. The findings of Dilek et al., (2016) indicated that the local activation of iNOS synthase was mediated by myeloperoxidase (MPO) of neutrophils in psoriasis, and thus the synthesis of molecules that target MPO or the synthesis of MPO in the lesion area may contribute to the development of a new treatment option.
The molecular docking analysis reveals that the bioactive molecules of SVK bind effectively with COX 2 and iNOS proteins, as evidenced by negative binding values. COX 2 and iNOS show the highest negative docking scores, indicating that proper interactions occur between the SVK ligands and the targets of the psoriasis pathway. These results are in agreement with the clinical practice of SVK and also with the existing literature in Siddha classical texts for the treatment of psoriasis.
The US Food and Drug Administration (FDA) has approved 117 distinct routes of administration for a drug molecule. Out of 117 different routes, oral administration is the most popular and easiest one (Zhong, 2017). Once a drug enters the body, the body starts to process the four aspects of a drug: absorption, distribution, metabolism (biotransformation), and elimination (commonly called ADME). QikProp is an advanced tool for predicting pharmacokinetic and physicochemical (ADME) properties of small organic molecules based on the full 3D molecular structure. The Swiss ADME web tool enables the computation of key physicochemical, pharmacokinetic, drug-like, and related parameters for one or multiple molecules. Both these online tools were used to assess the ADMET of various SVK ligands. Importantly, none of the molecules from SVK violated more than one of Lipinski’s Rule of Five criteria. This adherence to the rule is essential for evaluating drug-likeness and predicting oral bioavailability, thereby enhancing the potential for successful drug development. MDCK and CaCo-2 values suggest that the bioactive molecules from SVK possess strong capabilities for crossing both the blood–brain and gut barriers, highlighting their promising bioavailability and therapeutic potential.
Once a drug candidate has been successfully developed, conducting a comprehensive toxicity analysis becomes crucial. Detailed toxicity profiling has become essential to ensure the safety and efficacy of the bioactive molecules. Using the ProTox-I server, the toxicity profiles were meticulously estimated by evaluating key parameters, including hepatotoxicity (liver toxicity), carcinogenicity (cancer-causing potential), mutagenicity (genetic mutation potential), and cytotoxicity (cell damage potential). No hepatotoxicity, cytotoxicity, carcinogenicity, mutagenicity, or immunotoxicity were reported for SVK ligands. This ensures the safety of SVK ligands, and these results are in agreement with their therapeutic usage by Siddha practitioners.
The multifaceted approach for treating psoriasis will initially address the inflammation, immune response, and skin regeneration. From the present in silico study, it was clear that the primary goal of SVK mainly focuses on decreasing the inflammatory process, and this may in turn reduce the symptoms and complications. The drug efficiently manages the quality of life for patients by effectively reducing psoriasis symptoms. SVK also helps to speed up the healing process and the repair and regeneration of the skin barrier, aiding in faster recovery and reducing the severity of skin lesions.
5. CONCLUSION
SVK has been used for ages to treat a wide range of skin diseases, including psoriasis, in the Siddha system of medicine. The present study is the first of its kind to demonstrate the scientific validation of SVK usage in clinical practice. The GC-MS analysis reveals the abundance of the phytocomponents and validates the traditional usage of SVK for psoriatic ailments. Molecular docking studies revealed that SVK interacts with two target proteins, COX 2 and iNOS, and reduces their expression, which in turn decreases inflammation, a key step in psoriasis pathogenesis. Various ADMET tools and toxicity prediction tools also revealed the drug-likeness properties and lack of toxicity profiles of SVK ligands. Further elaborative studies are required to elucidate the mechanism of action of SVK in the treatment of psoriasis through various pre-clinical and clinical trials, especially randomized controlled trials (RCTs), to establish the scientific validation of this classical Siddha formulation for its global acceptance.
ACKNOWLEDGEMENT
The authors express their sincere gratitude to Director General, Central Council for Research in Siddha (Ministry of Ayush, Government of India) for his constant support and encouragement in research activities. The authors also acknowledge Dr. Manikandan Jayaraman, Project Scientist–1, Dept. of Bioinformatics, Alagappa University, Tamil Nadu, for his support in conducting the molecular docking study.
AUTHOR CONTRIBUTIONS
G.G.: Research concept and design, Data analysis and interpretation, Writing the article, Critical revision of the article, Final approval of the article; R.R.: Research concept and design, Data analysis and interpretation, Writing the article, Critical revision of the article, Final approval of the article; R.K.: Collection and/or assembly of data, Data analysis and interpretation, Writing the article, Final approval of the article.