

Introduction
The pandemic of Covid-19 is of an arising contagious affection, called coronavirus affection 2019 or Covid-19, caused by the coronavirus SARS-CoV-2 (Hui et al., 2020), which appeared in Wuhan City on November 17th, 2019, in central Chinese (Hubei Province), before spreading in numerous countries. The World Health Organization (WHO) first warnings the People’s Republic of China and its other member countries, and also declares a state of public health exigency of transnational concern on January 30th, 2020. On March 11th, 2020 (Chauhan, 2020), the new infection caused by this virus was confirmed as a pandemic by the WHO, which called for crucial protecting measures to prevent the overload of intensive care services and to reinforce precautionary hygiene avoidness of all physical contact kinds between people. Promotion of social distancing, and prohibition of crowds and major events as well as several travel restrictions, making people more aware about hand washing as a crucial step of preventive measures, implementation of quarantine, etc. In the whole world, countless cancellations of sport and cultural events was causing by this world-wide pandemic, several countries have put in place containment measures to slow down the appearance of new sources of contagion, even through the closure of their borders. Moreover, the spread of this pandemic had a negative impact on the social life of people as well as on the global economic stability since it has further slowed down the social and economic activities (Chauhan, 2020).
Currently, there are a little approved treatment that can treat the infection caused by SARS-CoV-2, consequently, there is an urgent claim for more chemotherapeutic agents to stop this disease.
Coronaviruses (CoVs) are single-stranded positive-sense RNA viruses that have enormous viral RNA genomes (V’kovski, Kratzel, Steiner, Stalder, & Thiel, 2020). Several works have shown that SARS-CoV-2 has a comparable genomic structure to that of beta-coronaviruses (Andersen, Rambaut, Lipkin, Holmes, & Garry, 2020; Chauhan, 2020). It consist of a 5′-untranslated region, a replicase complex that encode non-structural proteins, a spike protein gene, envelope protein gene, a membrane protein gene, a nucleocapsid protein gene, and numerous unidentified non-structural open reading frames (Islam et al., 2020; Qamar, Alqahtani, Alamri, & Chen, 2020). Therefore, and like other beta-coronaviruses, this virus can be terminated by blocking these proteins. Chloroquine, hydroxychloroquine, lopinavir/ritonavir, favipiravir, remdesivir, nitazoxanide, and ivermectin were the commonly used drugs, they have showed their efficacy in inhibiting SARS-CoV-2 (Yavuz & Ünal, 2020). Those agents can selectively and potently inhibit some nonstructural proteins like; the Main protease and Helicase (Nsp13) (Mpro) (Jin et al., 2020), Papain-like protease (PLpro) (Rut et al., 2020) and structure proteins like (Xia et al., 2020); protein S, protein E and protein M. Hence, the search for new molecules with certain inhibitory activity is very extensive and gaining interest of researchers to test new molecules. In many previous studies, bioactive molecules originated from natural resources have been proven to possess a potential antiviral activity. In the present study, the main idea is to focus on making a computational simulation of inhibition effects of some phenolic compounds using Protein-Ligand Molecular Docking approach. In recent years, the development of drugs through mathematical and computational approaches has become increasingly (Alamri et al., 2020; Bobrowski et al., 2020; Nukoolkarn, Lee, Malaisree, Aruksakulwong, & Hannongbua, 2008) .
Materials and methods
Proteins sequence preparation
For this work, four enzymes are chosen as targets:
Main protease (Mpro) downloaded from the PDB database encoded by 6LU7 (Jin et al., 2020) in a resolution of 2.16Å, is also called chymotrypsin-like protease (3CLpro). It is responsible for cleaving most of the polyprotein sites and the results are non-structural proteins (nsps) that come together in the replicase-transcriptase complex (RTC).
Papain-like protease (PLpro) taken from the PDB database with code of 6WUU (Rut et al., 2020) with a resolution of 2.79Å, cleaves the nonstructural protein 1-2, 2-3 and 3-4 boundaries and works with main protease to cleave the polyproteins into nsps.
Helicase (Nsp13) catalyses the unwinding of duplex oligonucleotides into single strands in an NTP-dependent manner. The SARS-CoV-2 helicase X-ray structure was built based on 6JYT (Jia et al., 2019) ccode in a resolution of 2.80Å. Therefore, the ADP binding site (ADP site), and the nucleic acids binding site (NCB site) were defined for small molecule docking.
RNA-dependent RNA polymerase (RdRP) downloaded from the PDB database with code of 7BV2 in a resolution of 2.50Å, catalyses the RNA replication from an RNA template (Venkataraman, Prasad, & Selvarajan, 2018). Specially, it catalysis the RNA strand complementary synthesis to give template of RNA. This contrasts with typical DNA-dependent RNA polymerases, which used by all organisms to catalyse the transcription of RNA from a DNA template. Thus, we defined two docking sites: the RTP site, and the RNA site.
Ligand database preparation
156 phenolic compounds and 9 antiviral drugs were downloaded in three-dimensional (3D) illustration and SDF structural data format from open database of National Institutes of Health (https://pubchem.ncbi.nlm.nih.gov/) (Kim et al., 2016). All these molecules are chosen by their reported antiviral activities against several viruses as following; African swine fever virus (ASFV) , Respiratory syncytial virus (RSV), Echovirus, Enterovirus A71 (EV-A71), influenza A virus subtype H1N1 (A/H1N1), Influenza A virus subtype H6N1 (A/H6N1), hepatitis C virus (HCV) , human immunodeficiency virus (HIV), human respiratory syncytial virus (hRSV), Herpes Simplex (HSV-1 & HSV-2), Porcine epidemic diarrhoea virus (PEDV), severe acute respiratory syndrome coronavirus (SARS-CoV) (Table S1, Appendix A Supplementary material).
Table 1
Binding scores (kcal/mol) of the top three phenolic compounds against each protein target
Docking protocol
The proteins structures were downloaded as a Protein database files and then transformed into Protein Data Bank Partial Charge (PDBQT) format via MGLTools software (Trott & Olson, 2010). Before docking, polar-hydrogen atoms were included, and gasteiger charges were processed (Kong et al., 2020). Autodock-vina was used carried out to the structure-based virtual screening; the docking box was defined as the center of native ligand coordinates with 30Å×30Å×30Å in length to include the residues of complete cavity. MGLTools was used to add hydrogens and prepare pdbqt files for proteins and ligands with an exhaustiveness level at 12. The 2D interaction was generated using the LIGPLOT+ software. The validation of the best generated model was done by analysis of Ramachandran plot generated in Ramachandran Plot server (https://zlab.umassmed.edu/bu/rama/).
Drug-likeness analysis by ADMET profiling
Absorption, distribution, metabolism, excretion, and toxicology (ADMET) were performed online by SwissADME tools (Daina, Michielin, & Zoete, 2017) (http://www.swissadme.ch/index.php). Drug-likeness can provide the possibility for a phenolic compounds to be developed as an oral drug with respect to bioavailability by the assessment of The Lipinski (Lipinski, Lombardo, Dominy, & Feeney, 1997), Ghose (Ghose, Viswanadhan, & Wendoloski, 1999), Veber (Veber et al., 2002), Egan (Egan, Merz, & Baldwin, 2000) and Muegge (Muegge, Heald, & Brittelli, 2001) rule’s.
Statistical analysis
Frequency distribution and heatmap were performed by orange software version 3.27.1 (Demšar et al., 2013), input data was generated from the (Table S1, Appendix A Supplementary material).
Results and discussion
Effect of phenolic class on docking scores
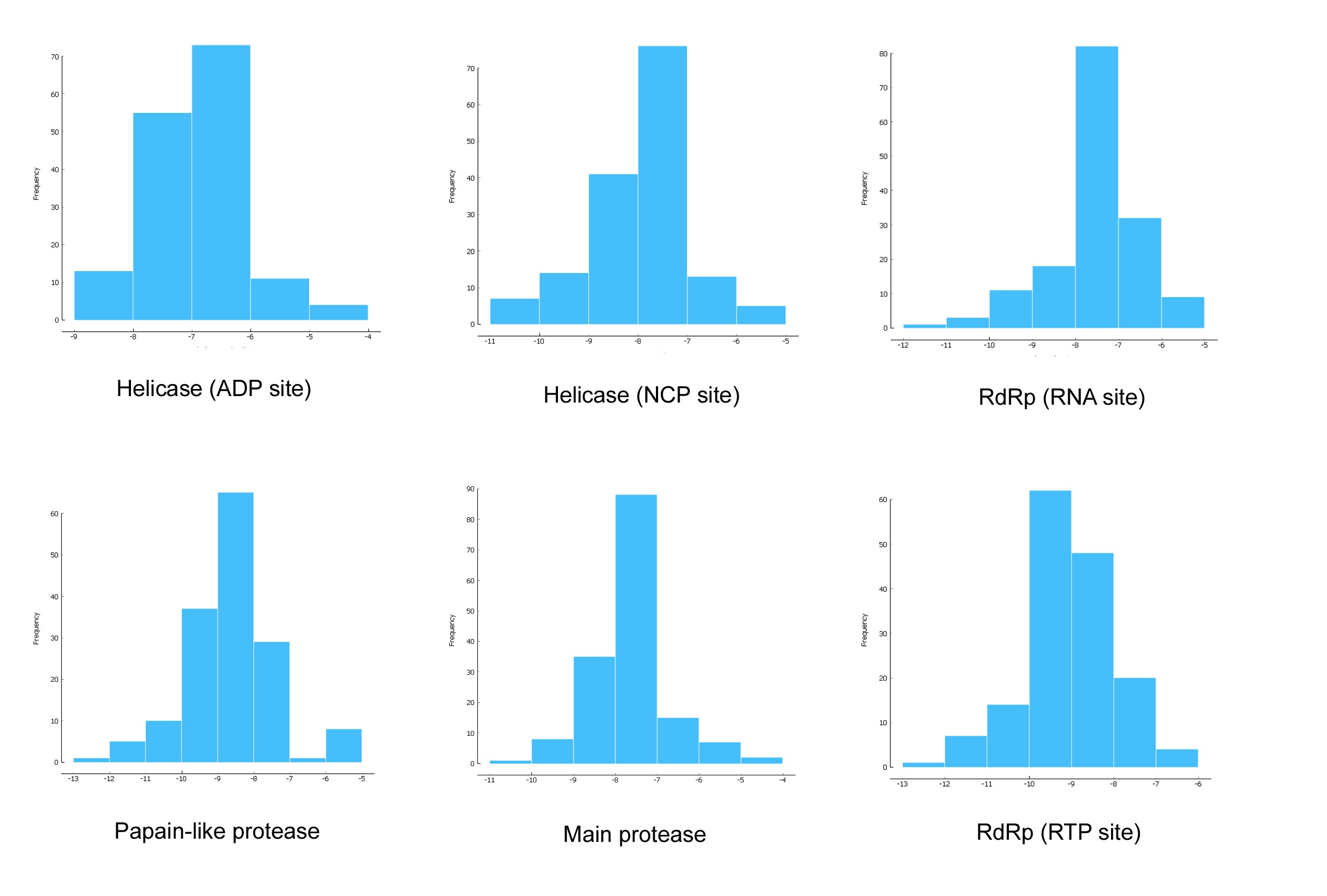
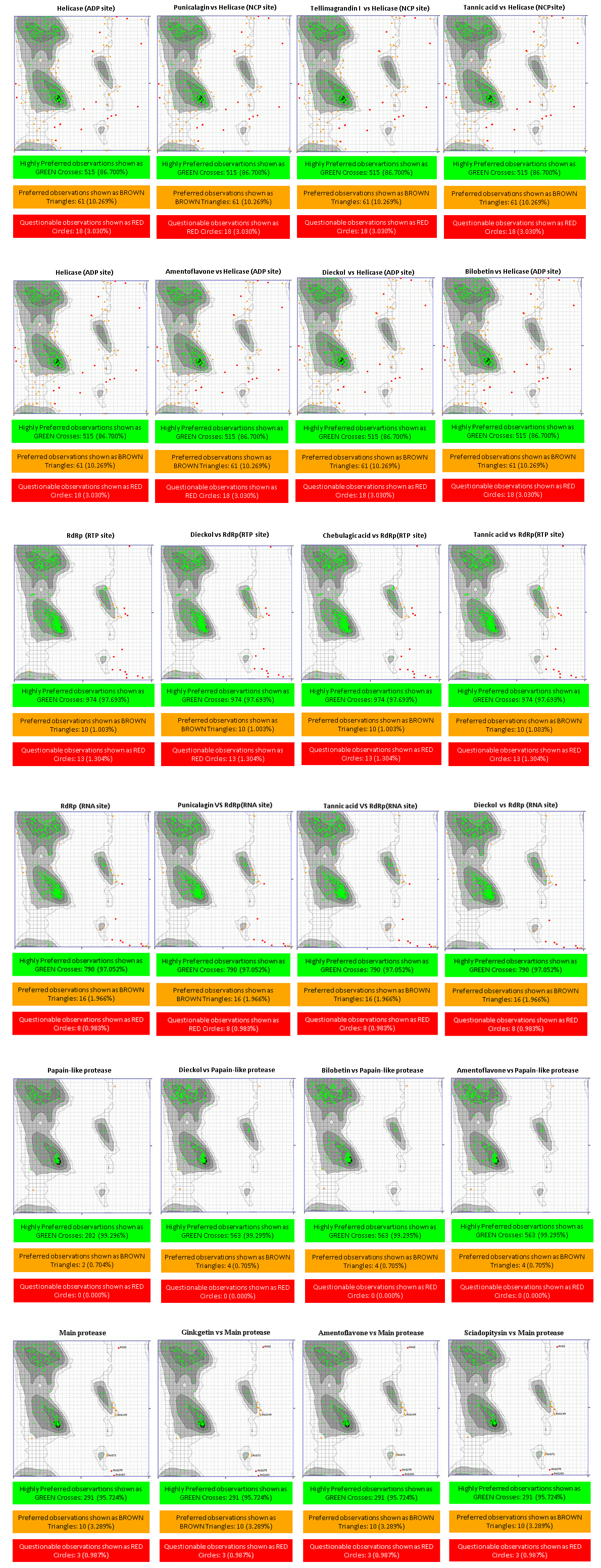
Figure 1 demonstrated that the distributions frequency of binding energy score among the different phenolic compounds and SARS-CoV-2 enzymes pre-selected varies between -5 and -12 kcal/mol. For Main protease, helicase (NCP site), RdRp (RNA site), the higher binding scores were recorded between -8 and -7 Kcal/mol with 56.41%, 48.72%, 52.56% respectively. As for the Helicase (ADP site), docking results show the lowest scores distributed with 46.79% between -6 and -7 Kcal/mol. 41.67% of binding scores were recorded at -8 and -9 Kcal/mol for papain like protease. Finally, the highest scores distribution characterize RdRp (RTP site) with 39.74% between -9 and -10 Kcal/mol. .
The result of a hierarchical k-means clustering (20 clusters) calculation is displayed in a heatmap (Figure 2) as a dendrogram, the row dendrogram shows the distance of similarity between rows, and which node belong to which row. As a result of clustering, Figure 2-A arranges the binding scores of ligands according to their phenolic classes (-5.011 and -12.60 kcal/mol), two principal clusters were manifested, the first (c1) regroups the high scores, the second regroups the low scores; ranging from -12 to -9 kcal/mol. Subcluster C1 generates four phenolic classes: tannins (Tannic acid, Tellimagrandin, Dieckol, Chebulagic acid, Pentagalloylglucose), biflavones (Amentoflavone, Bilobetin, Ginkgetin, Sciadopitysin), favan-3-ols (3-O-galloyl-procyanidin B2, Theaflavin) and flavonoid glycosides (Rhoifolin, Naringin, Neohesperidin, Hesperidin, Quercetin‐3‐O‐rutinoside, Diosmin, Kaempferol‐3‐O‐robinobioside, Kaempferol‐3‐O‐rutinoside). Subcluster C2 contains: Abietane, Anthocyanidin, Anthraquinone, Benzopyrans, Chalcones, Coumarins, curcuminoids, Flavanones, Flavanonols, Furanoflavonoids, Geranylated flavonoids, Glycosylated Lignans, Glycosylated Phenylpropanoids, Iridoid Glucoside, Isoflavanes, Isoflavanone, Isoflavones, Isoprenylated flavan, acid Phenols, Phenylflavonoids, Phenylpropanoids, prenylflavonoid, stilbenoids, xanthonoid.
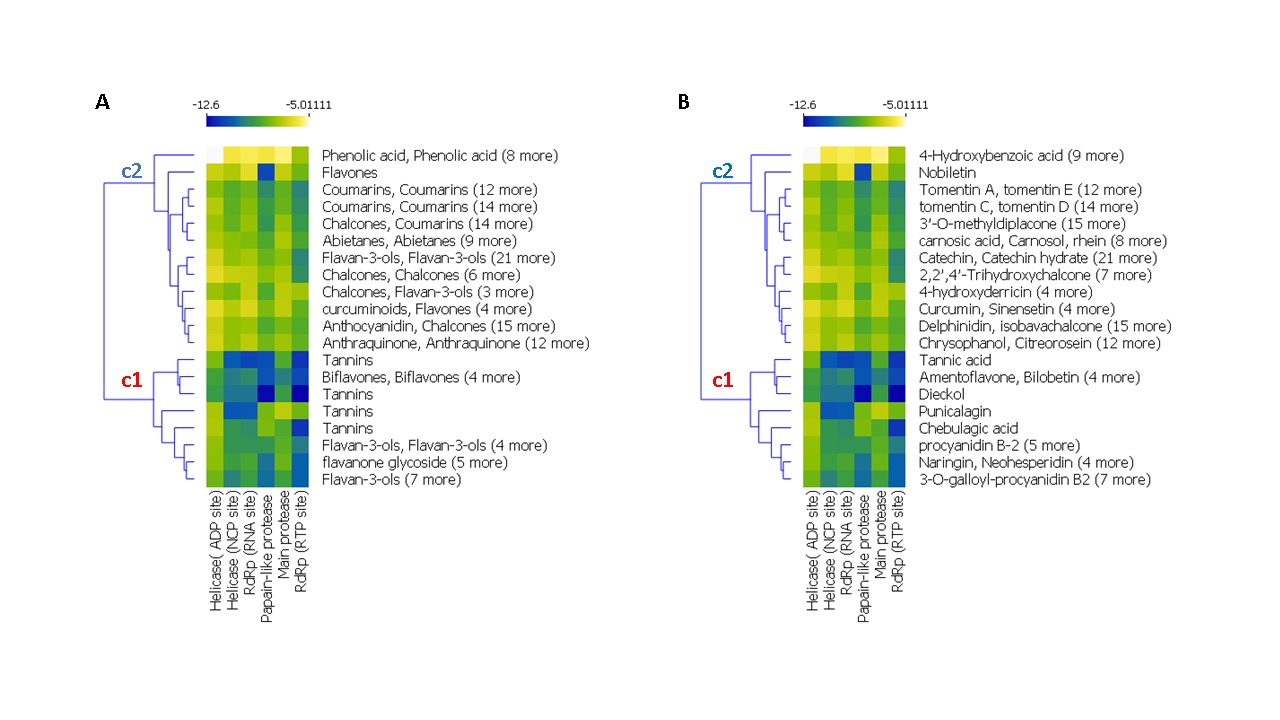
Table 2
Drug-likeness analysis of the top three molecules against each target of SARS-Cov-2
Phenolic acids show the highest binding scores close to -6 kcal/mol as an average. Consistently, previous docking simulations studies suggest the possibility that plant phenolic compounds can inhibit the responsible key factors r for the coronavirus life cycle (Russo, Moccia, Spagnuolo, Tedesco, & Russo, 2020). Chen et al. (2004) showed that the flavones glycosides have a potential anti-viral activity. In this work, ten SARS-CoV viruses were isolated from ten patients, Baicalin showed a half maximal effective concentration ranged from 12.50 to 25.00 μg/ml at 48 hours without significantly cell viability affection. In the same context, tetra-O-galloyl-β-d-glucose and luteolin, showed inhibitory activity for the entry process of SARS-CoV into host cells (Yi et al., 2004). These findings revealed that, biflavone and tannins class present an important binding energy score, which are somehow consistent with other empirical studies (Khalifa, Zhu, Mohammed, Dutta, & Li, 2020; Ryu et al., 2010). The outcomes of our study led us to go further steps; mainly, the study of binding profile and the correlation between amino acid’s proteins and proposed ligands.
Analysis of screened inhibitor interaction
To comprehend the binding mode of flavonoids interactions with SARS-CoV-2 enzymes, 156 phenolic compounds and 9 antiviral drugs were selected from literature. 9900 docking assays were conducted (including the replications to validate molecules binding energy scores). Following the ligands docking, many scores was established for all six-target proteins. The found results were set in rising score values order (Table S1, Appendix A Supplementary material). To simplify this work, the top three molecules against each target were selected (Table 1).
Main protease (Mpro as a target
Main protease (Mpro) is a potential pharmacological target against SARS CoV-2. It is a vital virus enzyme since it is indispensable for polyproteins proteolytic processing (Estrada, 2020). It is responsible for cleaving most of the polyprotein sites and the results are non-structural proteins (nsps) that come together in the replicase-transcriptase complex (RTC).
According to docking results (Table S1, Appendix A Supplementary material), the highest binding energy scores were recorded for biflavone, flavan-3-ols, flavonol glycoside, flavanone glycoside, flavonol glycoside, flavone glycoside, Tannins. 16.66% of tested ligands (26 phenolic compounds) have a score value superior to Remdesivir one used as a target control (Score Value: 8.4 kcal/mol), the top three ranked compounds against Mpro were compiled in Table 1.
Sciadopitysin has the best binding score (-10.1 kcal/mol), it is an active component that can be extacted from Taxus chinensis (Gu, Li, Chen, Yao, & Ou, 2013) , Ginkgo biloba (Liu et al., 2018), Torreya nucifera (Ryu et al., 2010). Sciadopitysin possess a large pharmacological activity such as phosphatase inhibitor of regenerating liver-3 (PRL-3) (Choi et al., 2006), inhibitor of the amyloid-beta (Aβ) peptide aggregation (Gu et al., 2013) . Scientific report of (Ryu et al., 2010) , demonstrated that biflavonoids from Torreya nucifera exhibited an important SARS-CoV 3CLpro inhibitory activity (62% at 100 μg/mL). Further, in our findings it was found that Sciadopitysin can realize 3 hydrogen bonds (HB) with Gly143(A): 3.18Å as a distance between residue and target, Thr26(A): 2.82Å, Thr26(A): 2.86Å and 13 hydrophobic interactions (HI); Glu166(A), Gln189(A), Arg188(A), Asp187(A), Met165(A), HIs164(A), His163(A), His41(A), Cys145(A), Leu27(A), Thr25(A), Asn142(A), Leu141(A). Similar studies on 3CLpro interaction with sciadopitysin revealed a high binding score: 9.1kcal/mol (Augustin, Hajbabaie, Harper, & Rahman, 2020) and -9.2 kcal/mol (Rana, Sharma, & Ghosh, 2020).
Amentoflavone, the second ranked ligand of Mpro with -10 kcal/mol binding score is a biflavone (bis-apigenin coupled at 8 and 3' positions, or 3′,8′′-biapigenin) originated from several plants including Ginkgo biloba (Lobstein-Guth, Briancon-Scheid, Victoire, Haag-Berrurier, & Anton, 1988), Hypericum perforatum (Michler, Laakmann, & Wagner, 2011), Xerophyta plicata (Williams, Harborne, & Tomas-Barberan, 1987). and Chamaecyparis obtusa (Krauze-Baranowska, Pobłocka, & Helab, 2005). Recently, it is proved to possess interesting activities, including protective antioxidant effects (Li, Chen, Niu, Zhou, & Li, 2020) and cyclooxygenases inhibitor (Banerjee, Valacchi, Ziboh, Der, & Vliet, 2002). In virology, it has been reported that amentoflavone could reduce coxsackievirus B3 replication (Wilsky et al., 2012). Also, it showed strong antiviral activity against HSV-1 and ACV-resistant strains (Li et al., 2019). Results obtained by ligplot software disclosed that this biflavone interacts with Mpro protein through 3 hydrogen bonds: His163(A): 3.16Å, Thr26(A): 3Å, Thr26(A): 3.08Å, and 14 hydrophobic interactions: His41(A), Gln189(A), Arg188(A), Gln166(A), Met165(A), Asp187(A), His164(A), Leu141(A), Ser144((A), Thr25(A), Gly143(A), Leu27(A), Cys145(A), Ans142(A). Rameshkumar et al. (2021) reported that amentoflavone exhibited a binding affinity with a score of −8.1 kcal/mol against main protease. Consistently, additional work done on biflavonoids of Torreya nucifera leaves showed that amentoflavone had −9.2 kcal/mol binding affinity (Ghosh, Chakraborty, Biswas, & Chowdhuri, 2020).
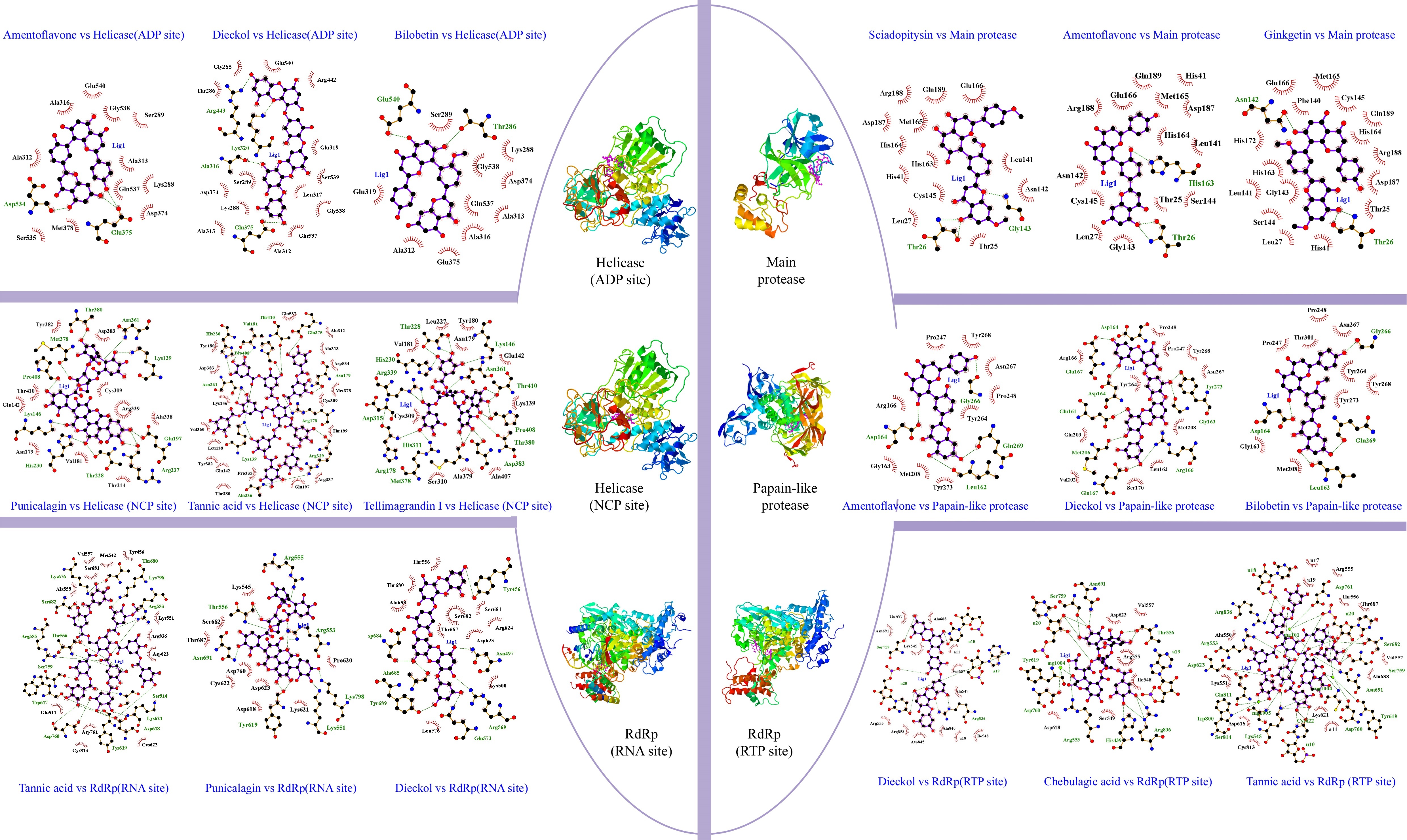

Ginkgetin a biflavonoid derived from leaves of Ginkgo biloba (Son et al., 2005), Selaginella moellendorffii (Sun, Syu, Huang, Chen, & Ou, 1997), Taxus chinensis (Ruan et al., 2014), ginkgetin exhibit numbers of pharmaceutical activities (Adnan et al., 2020) including antiinflammatory, antibacterial, leishmanicidal and antiplasmodial agent, antifungal, and antitumor activities (Lou, Bi, Chan, Dong, & Tsim, 2017). This bioflavonoid is the third best ligand with -9.8 kcal/mol, can realize 3 HB; Asn142(A): 2.78Å, Thr26(A): 3.11Å, Thr26(A):3.05Å and 16 Hydrophobic interactions; Met165(A), Cys145(A), Gln189(A), His164(A), Arg188(A), sp187(A) , Thr25(A), His41(A), leu27(A), Ser144(A), Leu141(A), Gly143(A), His163(A), His172(A), Glu166(A), Phe140(A). These present findings are concordant with the work of Rana et al. (2020) reporting that ginkgetin interact with main protease and have a potential docking binding score (-10.19 kcal/mol). There are 8 residues are in common shown in the Wenn diagram (Figure 5); Hr26(A), His41(A), Gln189(A), Arg188(A), Met165(A), Leu141(A), Gly143(A), Cys145(A). All the residues were confirmed with amino acid listed in Main protease active site (Table S2, Appendix A supplementary data). Data presented in this table, show that the phenolic compounds can occupy the predicted active site of the protein Mpro, and the stability of the protein-ligand complexes is ensured by the comparison of the stereochemical structures of these molecules by carrying out by Ramachandran Plot (Figure 4). The latter is used to validate the protein structure based on the φ (phi), ψ (psi) and ω (omega) angles values. The plots revealed that the structure of the protein remains intact even in complex with the three molecules. The RMSD of backbone alpha carbon atoms of Amentoflavone-MainPro, and ginkgetin main protease complex were reported recently by (Ghosh et al., 2020) . The average RMSD values (computed from five independent analysis) for unligated Mpro, Mpro-amentoflavone, and Mpro-ginkgetin complexes were found to be 0.297nm, 0.248nm and 0.246nm, respectively, suggesting that these two ligand-complexes were stable.
Papain-like protease (PLpro as a target
The papain-like protease (PLpro) is an interesting antiviral target because they are essential for the replication of coronaviruses (Shin et al., 2020), PLPro is a protease located in NS3 of the viral polypeptide, in addition, PLpro remove ubiquitin and ISG15 from the proteins of the host cell to help viruses evade innate host immune responses. Following a docking analysis of nine commercialized drugs, results revealed that Indinavir exhibited a higher binding score (-10.3 kcal/mol) for that it has been selected as the best control ligand. Fifteen phenolic compounds have a superior score values and the top three ranked were Dieckol, Bilobetin, Amentoflavone with score value: -12.5, -11.3 and -11.2 kcal/mol respectively (Table 1).
Dieckol, a phlorotannin, extracted from brown seaweed; Eisenia bicyclis (Koirala, Jung, & Choi, 2017), Ecklonia cava (Moon et al., 2011). It is known to act as antibacterial activity (Choi et al., 2014), UVB-photoprotective activity (Guinea, Franco, Araujo-Bazán, Rodríguez-Martín, & González, 2012), anti-inflammatory (Sanjeewa et al., 2020) and cytoprotective agents (Lee, Kim, Yoo, & Kwon, 2013). The antiviral activities of dieckol have been largely reported. Such as, in vitro antiviral activity of this active compound against murine norovirus (MNV) in RAW 264.7 cells has recorded EC50 of 0.90 µM (Eom et al., 2015). Additionally, it possesses an important activity against Influenza A viruses; [H1N1], [H9N2] and [H3N2] (Ryu et al., 2011). Park et al. (2013) demonstrated that dieckol showed significant inhibitory against SARS-CoV 3CLpro cell-free cleavage (IC50= 2.7 μM). These previous results encourage us to consider it as a potential drug candidate. Docking results revealed that this natural phlorotannin interacted through 8 HB; Asp164(C): 3.1Å, Glu167(C): 3.12Å, Asp164 (A): 2.69Å, Glu161(A): 2.83Å, Met206(C) 3.21Å, Glu167(A): 2.71Å, Arg166 (C): 3.22Å, Gly163(A):3.13Å, Tyr273(A): 2.7Å, Moreover, this compound also formed a large hydrophobic interactions network with the surrounding amino acids, including 11 residues; Pro248(A), Pro247(C), Tyr268(A), Asn267(A), Met208(C), Leu162(A), Ser170(C), Val202( C), Glu203(C), Tyr264(A), Arg166(A). Gentile et al. (2020) reported that dieckol was able to bind and inhibit the 3CLpro due to an extensive network of HB (binding score: -12 kcal/mol).
Bilobetin is a biflavone extracted from Ginkgo Biloba L plants. Recently it was reported to present several biological activities such as an antiviral (Freitas et al., 2009), anticancer (Li et al., 2219) , antinflammatory (Li et al., 2019) , antibacterial (Woldemichael, Singh, Maiese, & Timmermann, 2003) and antifungal (Krauze-Baranowska & Wiwart, 2003). Moreover, several computational works showed that bilobetin is a vigorous drug candidate for the treatment of herpes simplex virus (HSV) and hepatitis B virus (HBV) infection (Ramaiah & Suresh, 2013). According to results summarized in Table 1, it was found that the estimated docking binding score for bilobetin was (-11.3kcal/mol). Based on this value, we have further extended our study to check the interactions network between 3CLpro and bilobetin, Gly266(A): 2.89Å, Asp164(A): 3.04Å, Gln269 (A): 3.02Å, Leu162(A): 2.95Å represent the residues that interact with our ligand by hydrogen bonding, Asn267(A), Pro248(A), Thr301(A), Pro247(C), Tyr264(A), Tyr268(A), Tyr273(A), Met (208) C, Gly163 (A) were the amino acid having hydrophobic interactions with bilobetin. In similar study on PLpro (PDB ID: 6W9C) (Rana et al., 2020), it has been consistently shown that bilobetin could interact with Plpro protein through 7 HB with the following residues: Phe 140, Glu166, Gln 189, Thr 190 and Gln 192. It could also form 3 hydrophobic interactions (Met 165, Glu 166 and Pro 168), recording a freen energy binding score of -10.83 Kcal/mol.
Amentoflavone, as described before, showed the third potent affinity to 3CLpro (-11.2 kcal/mol), the 2D representation of ligand-protein binding including HB and HI are presented in Figure 3. Thirteen residues of 3CLpro interacted with the ligand, 4 HB were formed between Amentoflavone and Gly266(A): 3.01Å, Gln269(A): 2.85Å, Leu162(A): 2.82Å, Asp164(A): 3.19Å, while, 9 amino acid residues, Tyr268(A), Asn267(A), Pro248(A), Tyr264(A), Tyr273(A), Met208(C), Gly163(A), Arg166(C), Pro247(C), formed hydrophobic interactions. This result was approved by a recent work, elucidating by SARS 3C-like protease inhibition assay and molecular docking study that amentoflavone was the most potent SARS-CoV 3CLpro inhibitor with IC50 = 8.3 μM (Abdillah & Cita, 2020). Moreover, the potential of the inhibitor amentoflavone correlated well with binding energies −11.42 kcal/mol, In the former, the interaction of this biflavone with the substrate-binding pocket of 3CLpro involved 5 hydrogen bonds with: His163 (3.154Å), Leu141 (2.966Å), Gln189 (3.033Å), Val186 (4.228Å) and Gln192 (3.898Å). Wenn diagram summarize the intersection between catalytic residues interactions, (Figure 5), Asp164 (A) formed hydrogen bonds with the three ligands studied in this work. Leu162(A), Tyr268(A), Asn267(A), Pro248(A), Tyr264(A), Tyr273(A), Met208(C), Gly163(A), Pro247(C) formed Hydrophobic interactions. All the residues were confirmed with amino acid listed in 3CLpro active site (Table S2, Appendix A supplementary data). Highly Preferred observations shown in Ramachandran Plot (Figure 4) revealed that the interaction of ligands/enzymes do not affect the protein stereochemistry, which allows us to conclude that these complexes are stable and robust. In conclusion, of the results obtained, papain like protease can be considered as a potential target for SARS-CoV 2 inhibitors.
Helicase (Nsp13 as a target
The helicase is known as an enzyme that catalyze the NTP-dependent unwinding of duplex oligonucleotides into single strands (Kadaré & Haenni, 1997). Two sites for molecules binding: ADP site and NCB site were defined (Gupta et al., 2020). Here, three phenolic compounds were chosen as ligands for each binding site. Ritonavir (-9.4 kcal/mol) and Ivermectin (-9,7 kcal/mol) are respectively the drug’s bending energy scores for ADP and NCP site. As shown in Figure 2-A, Tannins, biflavones, flavonol glycosides are the most ranked ligands with SARS-CoV 2 Helicase.
ADP site docking revealed that amentoflavone (-9 kcal/mol), dieckol (-9 kcal/mol), bilobetin (-8.9 kcal/mol) were the most efficient ligands (Table 1). Until the time of writing this work, any docking model has not yet evaluated these molecules. The present results reveal that the ADP site interacts with amentoflavone by 3 HB; Asp534 (A): 3.72Å, Glu375 (A): 3.11Å, Glu375 (A): 3.15Å and 11 HI; Ala316 (A), Glu540 (A), Gly538 (A), Ser289 (A), Ala313 (A), Gln537 (A), Lys288 (A), Asp374 (A), Met378 (A), Ser535 (A), Ala312 (A). Dieckol formed 4 HB; Arg443(A): 3.21Å, Lys320(A): 3.02Å, Ala316(A) 2.89 Å, Glu375(A): 3.05Å and 14 HI; Thr286(A), Gly285(A), Glu540(A), Arg442(A), Glu319(A), Ser539(A), Leu317(A), Gly538(A), Gln537(A), Ala312(A), Ala313(A), lys288(A), Asp374(A), Ser289(A). Bilobetin interacts with the ADP site by 2 HB; Glu540 (A): 2.86Å, Thr286 (A): 3.25Å and 10 HI; Ala316 (A), Gly538 (A), Ser289 (A), Ala312 (A), Gln537 (A), Lys288 (A), Asp374 (A), Glu375 (A), Glu319 (A), Ala313 (A) (Figure 3).
Consistent report (Jia et al., 2019) demonstrated that the helicase NTPase activity exist in a split located between 1A and 2A motor domains and made by the following 6 amino acids; Arg567, Gln404, Glu375, Asp374, Ser289 and Lys288. Four basic amino acid residues (Arg337, Arg339, Lys345 and Lys347) located at the top of nucleic acid binding channel are critical for reducing helicase activity.
Four of Amentoflavone complex residues are conformed by this study, which means that our docking model can reveal a valid therapeutic approach. The nucleic acids binding site (NCB site) of helicase has more interesting binding energy scores than ADP site. The top three ranked molecules are Punigalagin, Tellimagrandin I and tannic acid with -10.9, -10.9 and -10.8 kcal/mol respectively. All these molecules are derivatives of natural tannins class, they are characterized by their high ability to reach a stable cross-linked association within different proteins by forming hydrogen bonding interaction (Fraga-Corral et al., 2020). Results in Figure 3 show that the best ranked hydrolysable tannins are pedunculagin, where it interact by 11 hydrogen bonds with SARS‐CoV‐2 Helicase (Nsp13) residues; Thr380(A): 2.34Å, Asn361(A): 2.78Å, Lys139(A): 2.72Å, Glu197(A): 2.76Å, Arg337(A): 2.93Å, Arg337(A): 3.13Å, Thr288(A): 2.93Å, His230(A): 2.98Å, Lys149(A): 2.98Å, Pro408(A): 2.85Å, Met378(A): 3.31Å. In addition, pedunculagin is stabilized within the NCP site pocket with 10 hydrophobic interactions, Asp383(A), Cys309(A), Arg339(A), Ala338(A), The214(A), Val181(A), Asn179(A), glu142(A), Thr410(A), Try382(A). The pedunculagin / Helicase interaction has not been studied by subsequent work, through molecular docking analyses, a recent study shows that pedunculagin are the best hydrolysable tannins that can interact with the catalytic site SARS‐CoV‐2‐3CLpro residu (Khalifa et al., 2020). Tito et al. (2020) suggested that a pomegranate peel extract, containing 182.31 mg Pedunculagin /g of dried peels powder, can inhibit the the 3CL protease activity until 80%, reduce the Angiotensin-converting enzyme 2 and Transmembrane protease serine 2 precursor gene expression level by 30% and 70% respectively and inhibit the interaction between S protein and Angiotensin-converting enzyme 2 until 74%. Another structurally similar molecule, Tellimargrandin I which is found widely in Cornus canadensis (Lavoie et al., 2017), Rosae Rugosae (Tamura et al., 2010) and Eucalyptus globulus (Boulekbache-Makhlouf et al., 2010) plants, was reviewed by Zheng et al. (2012) for its wide spectra of biological activities. Here, it recorded a binding score energy of −10.9 kcal/mol, This molecule was reported to form a large H bond network with Lys146(A): 2.88Å, Asn361(A): 3.08Å, Thr410(A): 3.03Å, Pro408(A): 3.07Å, Pro408(A): 3.07Å, Thr380(A): 3.01Å, Asp383(A): 3.12Å, Met378(A): 2.69Å, Met378(A): 3.11Å, Arg178(A): 3.07Å, His311(A): 3.07Å, Asp315(A): 2.96Å, Arg339(A): 3.13Å, Arg339(A): 2.94Å, His230(A): 2.68Å, Thr288(A): 3.32Å, Thr288(A): 3.16Å, Thr288(A): 2.75Å. Also, it ensure the binding mode by the intraction through 10 HI; tur180(A), Asn179(A), Glu142(A), Lys139(A), Alu407(A), Ala379(A), Ser310(A), Cys309(A), Val181(A), Leu227(A) (Figure 3). In related work, Puttaswamy et al. (2020) reported that Tellimargrandin I showed its ability of a robust interaction with Spike protein (-8.1 kcal/mol), RdRp (-9.5 kcal/mol), TMPRSS2 (-8.6 kcal/mol). Tannic acid is the third best ligand recording a binding score energy of -10,8 kcal/mol, it can realize 16 hydrogen bonds; Glu375(A): 2.75Å, Asn179(A): 3.07Å, Arg179(A): 3.01Å, Arg339(A): 2.89Å, Arg339(A): 2.87Å, Ala336(A): 3.05Å, Ala336(A): 3.07Å, Lys139(A): 3.25Å, Lys139(A): 3.25Å, Asn361(A): 2.88Å, Asn361(A): 2.91Å, His230(A): 2.83Å, Val181(A): 3.01Å, Val181(A): 3.26Å, Pro408(A): 3.10Å, Thr410(A): 3.11Å, and 13 hydrophobic interactions; Gln537(A), Ala312(A), Ala313(A), Asp534(A), Met378(A), Cys309(A), Thr199(A), Arg337(A), Gln197(A), Pro335(A), Thr380(A), Gln142(A), Leu138(A), Tyr382(A), VAl360(A), Lys146(A), Asp383(A), Tyr18(A) (Figure 3). Tannic acid is polyphenolic compound with high molecular weight, highly soluble in water. shown to possess antioxidant (Gülçin, Huyut, Elmastaş, & Aboul-Enein, 2010), antimutagenic (Chen & Chung, 2000) and anticarcinogenic properties (Baer-Dubowska, Szaefer, Majchrzak-Celińska, & Krajka-Kuźniak, 2020). It has been stated to present the activity against Influenza A virus, Papilloma viruses, noroviruses, Herpes simplex virus type 1 and 2, and human immunodeficiency virus (HIV) (Kaczmarek, 2020). Moreover, in 2005, Chen et al. (2005) reported in in vitro study that tannic acid, was the potent molecule to inhibit the SARS-CoV-2 3CLpro with high activity expressed by a low IC50 value (3 µM).
Wenn diagram (Figure 5) summarizes the intersection between catalytic residues interactions. Ala316 (A), Glu540 (A), Gly538 (A), Ser289 (A), Ala313(A), Gln537(A), Asp374(A), Ala312(A) for Helicase (ADP site), Thr380(A), Asn361(A), Lys139 (A), His230 (A), Pro408 (A), Met378 (A), Asp383 (A), Cys309 (A), Arg339 (A), Val181 (A), Asn179 (A) and Thr410 (A) represent the common amino acids for Helicase (NCP site). All the residues were confirmed with amino acid listed in Helicase active site (Table S2, Appendix A supplementary data), leading us to conclude that these complexes are made in the right way. Ramachandran Plot (Figure 4) distributes the highly preferred observations, revealing that the protein stereochemistry is not affected by ligands/enzymes interaction, which confirms that these complexes are stable and robust. Taken together the obtained results, we can conclude that Helicase could be considered as a prospective target for covid virus inhibitors.
RNA-dependent RNA polymerase (RdRP
According to docking results (Table S1, Appendix A supplementary data), for RNA site the most binding energy scores were recorded for biflavone, flavonol glycoside, Tannins classes. Eight phenolic compounds have a scores superior to Ritonavir one used as a target control (Score Value: -9.5 kcal/mol). The top three compounds ranked against RNA site (Tannic acid, punicalagin, dieckol) were compiled in Table 1. Concerning RdRp (RTP site), Rupintrivir (-10,8 kcal/mol) was used as a target control, 13 phenolic compounds have a scores superior to this control. The top three ranked compounds against RdRp (RTP site); Dieckol, Chebulagic acid and tannic acid, were compiled in Table 1.
As for the SARS-CoV-2 RdRp (RNA site), Pedunculagin was recorded as the best ligand among 156 tested phenolic compounds against SARS-CoV-2 RdRp (RNA site). The origin and well-made of Punicalagin are already cited in helicase section, Binding interactions of the native ligand (binding score = -10.8 kcal/mol). As shown in Figure 3, there are 11 hydrogen bonds with Arg555(A): 2.85Å, Arg553(A): 3.16Å, Arg553(A): 3.11Å, Arg553(A): 2.94Å, Lys798(A): 3.08Å, Lys551(A): 3.15Å, Tyr621(A): 2.93Å, Tyr621(A): 2.73Å, Asn691(A): 3.06Å, Thr556(A): 3.03Å, Thr556(A): 2.77Å. Additionally, other interactions were observed with Pro620(A), Lys621(A), Asp618(A), Asp623(A), Cys622(A), Asp760(A), Thr687(A), Ser682(A), Lys545(A). The second ranked ligand is tannic acid (binding score = -10,6 kcal/mol) with 32 ligand/protein interactions (Figure 3); 20 of them were hydrogen bonds, Arg553(A): 2.91Å, Arg553(A): 2.83Å, Thr556(A): 2.77Å, Thr556(A): 3.03Å, Thr556(A): 2.80Å, Lys798(A): 3.00Å, Ser682(A): 3.16Å, Lys676(A): 3.00Å, The680(A): 2.87Å, Arg555(A): 2.96Å, Ser814(A): 2.84Å, Trp617(A): 2.90Å, Ser759(A): 2.69Å, Asp760(A): 2.88Å, Asp760(A): 3.28Å, Tyr619(A): 2.73Å, Lys621(A): 2.94Å, Asp618(A): 3.01Å, Asp618(A): 2.78Å, Asp618(A): 2.82Å, and 12 Hydrophobic interactions, Arg836(A), Cys622(A), Tyr456(A), Asp623(A), Lys551(A), Ala558(A), Val557(A), Ser681(A), Met542(A), Asp761(A), Cys813(A), Glu811(A). As described before, Dieckol is a major marine polyphenol reported for its wide antiviral activity, exhibited a docking score of -10.1 kcal/mol. Ligand interaction analysis of the Dieckol /RdRp (Figure 3) complex shows that ligand mostly made 7 hydrogen bonds; Tyr456(A): 2.79Å, Asp684(A): 3.11Å, Ala685(A): 3.20Å, Tyr689(A): 3.02Å, Gln573(A): 2.97Å, Arg569(A): 3.17Å, Asn497(A): 3.03Å and 10 hydrophobic interactions; Thr556(A), Thr680(A), Ala688(A), Leu576(A), Lys500(A), Thr687(A), Asp623(A), Ser682(A), Arg624(A), Ser681(A). The three ligands studied in this section are tannins, this result leads us to investigate the ability of this phenolic class to inhibit RNA dependent RNA polymerase. Wenn diagram summarizes the intersection between residues interactions, (Figure 5), Thr556 (A), Asp623 (A), Ser682 (A) were the common residue. Moreover, these amino acids were listed in the (Table S2 Appendix A supplementary data) and cited as interactive residue in several works (Ahmad, Ikram, Ahmad, Rehman, & Mushtaq, 2020), in SARS-CoV-2 RNA Dependent RNA polymerase (RdRp) docking study, Ahmad et al. (2020)Ahmad et al. (2020) showed that Thr556, Asp623 appear among the amino acids that interact with Carbetocin and Ser682 with Colistin, Demoxytocin and Lanreotide, with an interesting docking score. Further, 3-O-alpha-L-arabinopyranosyl-echinocystic acid and Genkwanin 8-C-beta-glucopyranoside were reported as potent RNA-Dependent RNA-Polymerase inhibitors involving Thr556 and Asp623 as hydrogen bonding residues (Khan et al., 2020). According to the obtained results, it could be concluded that probably the trio Thr556 (A), Asp623 (A), Ser682 (A) can be a catalytic triad for RNA-Dependent RNA-Polymerase. The binding mode stability was approved by the comparison of the backbone conformation of native SARS-COV-2 enzyme and the different molecules. Ramachandran plot showed that the residues were present in the favoured regions and the protein structure remains intact even in complex with the three molecules.
For the RdRp (RTP site), Rupintrivir was the best FDA-approved antiviral drug with binding score -10.8 kcal/mol, 15 phenolic compounds were found having superior scores. The major part of these molecules were tannins, biflavones, flavanone glycosides, flavan-3-ols and flavonolignans. The top-ranked ligands are summarized in Table 1. Dieckol had the lowest binding energy score, -12.6 kcal/mol. The molecules were found to have 21 interactions (Figure 3); 3 HB with native RdRp (Ser759(A) 3.30Å, Arg836(A) 2.99Å and 3.08Å), 5 HB with RNA (u10(T) 3.01 and 3.03Å, a19(P) 3.05Å, u20(P) 3.07 and 2.69Å), 11 HI were also recorded, 2 with RNA (a11(T), U18(P)) and 9 with RdRp residues (Ala688(A), Thr687(A), Asn691(A), Val557(A), Ala547(A), Ile548(A), Asp845(A), Arg858(A), Arg555(A), Lys545(A)). The second ranked ligand is Chebulagic acid, it is a benzopyran tannin found in Terminalia chebula (Han, Song, Qiao, Wong, & Xu, 2006), It has been proved to be immunosuppressive (Hamada et al., 1997), antitumor agent (Wang, Li, & Hu, 2018), hepatoprotective (Kinoshita, Inoue, Nakama, Ichiba, & Aniya, 2007), and alpha-glucosidase inhibitor (Sasidharan et al., 2012). In 2011, Lin et al. (2011) identified chebulagic acid and punicalagin, two hydrolysable tannins from fruits of Terminalia chebula Retz, which exhibit their antiviral activities by targeting HSV-1 viral glycoproteins that interact with cell surface heparin sulphate. In 2013, Lin et al. (2013) confirmed the antiviral activity of these tannin by exploring the antiviral potential of these two tannins against several viruses that use glycosaminoglycans for entry. The ligplot results (Figure 3) show that this chebulagic acid interacts with RdRp with 17 hydrogen bonds; 12 HB with native RdRp (Asn691(A): 3.33Å, Ser759(A): 3.23Å, Ser759(A) : 3.11Å, Arg553(A) : 2.97Å, His439(A) : 3.13Å, Arg836(A) : 3.19Å, Arg836(A) : 2.98Å, Arg836(A) : 3.07Å, Thr556(A) : 2.78Å, Thr556(A) : 2.70Å), 2 HB bonded to magnesium of the protein (Tyr619(A) 3.34Å and Asp760(A) 3.17Å) and 5 HB with RNA (u20(P) 3.06Å, u20(P) 3.11Å, u20(P) 2.95Å, a19(P) 2.84Å). Furthermore, this compound formed a hydrophobic interactions network with the surrounding amino acid, including 11 residues Val557 (A), Asp623 (A), Arg555 (A), Ile548 (A), Ser549 (A) and Asp618 (A). Tannic acid is the third best ligand with -11.7 kcal/mol, it can realize 28 HB and 13 HI, as for chebulagic acid, the hydrogen bonds are distributed in three categories: 11 RNA/ ligands, 11 RdRp/ ligands and six RdRp/ magnesium /ligands. The 11 RNA/ ligands are reported with u20(P), u10(T) and u18(P) having different distances, u20(P): 2.33Å, 3.03Å, 3.13Å,2.90Å, 2.82Å, 2.74Å, u10(T): 2.85Å, 2.85Å, 3.02Å and u18(P) 2.78Å, 2.98Å, the 11 RdRp/ ligands are Asn691(A) 3.03Å, Ser759(A) 2.70Å, Cys622(A) 3.06Å, Lys545(A) 3.11Å, Lys545(A) 3.04Å, Lys545(A) 2.92Å, Ser814(A) 2.81Å, Asp623(A) 2.68Å, Arg553(A) 2.76Å, Arg836(A) 2.94Å, Ser682(A) 2.92Å. The 6 RdRp/ magnesium/ligands are Asp761(A): 2.33/mg101(P)/3.08, Asp761(A): 2.63/mg101(P)/3.08, Trp800(A): 3.18/mg1005(A)/2.38, Glu811(A): 3.09/mg1005(A)/2.66, Tyr619(A): 3.34/mg1004(A)/2.61, Asp760(A): 3.17/mg1004(A)/3.19. Moreover, Tannic acid is stabilized by 13 HI, Lys551(A), Ala550(A), Arg555(A), Thr556(A), Thr687(A), Val557(A), Ala688(A), Lys621(A), Cys813(A), Asp618(A) with RdRp and a11(T), u17(P), a19(P) with RNA (Figure 3). This mass of interactions is explained by the fact that tannic acid has many hydroxyl groups and a very variable geometric structure. Wenn diagram (Figure 5) summarizes common residues and nucleotides. It was found that the three ligands interact with u20 (P), a19 (P) from the RNA nucleotides and with Asn691 (A), Ser759 (A), Arg836 (A), Val557 (A), Arg555 (A) from the RdRp residues. These findings are in agreement with the work ofSingh, Pathania, and Rawal (2020) who revealed that Remdesivir (an FDA-approved intravenous antiviral drug) characterized by HB interaction with u20 from RNA nucleotides and having a binding pocket residue including Arg553, Arg555, Thr556 and Asn691, explaining its potential inhibitory effect. Moreover, Bastikar, Bastikar, and Chhajed (2020) confirmed that the docking of curcumin and allicin derivatives with RdRp exhibited notale binding affinity and having interactions with U10, A11 and U20 nucleotides and amino acid residues such as Asp623, Asn691, Arg555 and Ser682 which are highly involved in the HB with most of the tested ligands. Ramachandran Plot (Figure 5) distribute the highly preferred observations, which revealed that the protein stereochemistry is not affected by ligands/enzymes interaction, thus it is confirmed that these complexes are stable and robust.
From the obtained results, we can conclude that the molecular docking analysis could be a good tool for virtual screening and preliminary step towards searching for effective drugs against selected targeted protein/ enzyme.
Drug-likeness
Pharmacophore modelling to identify drug-like compounds is a common tool for in silico drug identification (Yang, 2010) by Drug-likeness analysis. SwissADME is a tool provided by the Swiss institute of Bioinformatics (Daina et al., 2017), used to predict physiochemical properties, lipophilicity, water-solubility, pharmacokinetics, drug-likeness and medicinal Chemistry. According the results of drug-likeness analysis predictions all the studied phenolic compounds have small bioavailability score. The Abbott bioavailability score (ABS) determines whether it passes or violates Lipinski’s rule of five. Generally, a molecule is estimated to have more than 10% of bioavailability (F) in case it passes Lipinski’s rule of five with ABS of 0.55 in pH constant conditions (Martin, 2005).
In current study, Chebulagic acid has reduced bioavailability (ABS: 0.11), Amentoflavone, Dieckol, Punicalagin, Tellimagrandin I, Tannic acid and Punicalagin recorded a bit higher score (ABS: 0.17), Bilobetin, Sciadopitysin, Ginkgetin revealed a relative high bioavailability (ABS: 0.55). Nevertheless, their bioavailability score makes them ineffectual via oral route. Similar results are recorded with FDA SARS-Cov-2 recognized drugs, they varied from 0.17 to 0.56 (Table S3 Appendix A supplementary data). Lipinski’s rule of 5 provides an overview of the drug-like inhibitor candidate (Lipinski et al., 1997). In general, phenolic compounds presenting a violation value higher than 1 do not meet the criteria for drug-likeness (MW less than 500, cLogP less than 5, number of hydrogen-bond acceptors (HBA) less than 10 and number of hydrogen-bond donors (HBD) less than 5). In fact, the study of our phenolic compounds shows only 3 candidates with one violation: Bilobetin, Sciadopitysin and Ginkgetin. The rest of molecules exhibited a higher MV than that proposed by Lipinski, but it is crucial to note that Ivermectin, Lopinavir, Remdesivir, Ritonavir, Rupintrivir, Tipranavir, the most probable drugs according clinical studies, present similar MW :875.09, 628.80, 602.58, 720.94, 598.66, 602.66 DA respectively (Table 2). Furthermore, according to Veber’s rules (Veber et al., 2002), satisfactory bioavailability is more suitable for compounds with ≤10 rotatable bonds (RB) and topological polar surface area (TPSA) ≤ 140 Å2. Considering these rules, the predicted proprieties point to that among nine drug-like inhibitor candidates, 8 compounds with TPSA > 140 Å2 fulfill the Veber’s rules and only Tannic acid with 31 RB do not fulfill the rules. PAINS (Pan Assay Interference Compounds) (Baell & Holloway, 2010) evaluation revealed that only Amentoflavone, Dieckol, Bilobetin, Sciadopitysin and Ginkgetin do not present any PAINS alert. Punicalagin, Tellimagrandin I, Tannic acid and Chebulagic acid present one alert, this distribution is a function of chemical class. In fact, biflavones do not present any resemblance to PAINS, but tannins can be classified as PAINS. This tendency is also observed with another medicinal chemistry model, biflavones were not determined to have any Brenk fragments (Brenk et al., 2008). Similar trend has been proved by previous study (Sayed et al., 2020), in which authors confirm that biflavones mainly amentoflavone have an efficient drug-like properties, based on their pharmacokinetics parameters (e.g., absorption and bioavailability), and Lipinski’s rules of 5 obedience (Table 2).
Conclusion
Taken together all the attained results, we can concluded that the screening of 9900 ligands / SARS-Cov-2 Proteins complex through docking analysis is a promising way to identify possibly effective drugs against COVID-19. By studying the interactions of 33 phenolic classes with six viral vital enzymes (Main protease, Papain-like protease, Helicase (ADP and NCP sites) and RNA-dependent RNA polymerase (RNA and RTP sites)). biflavone and tannin classes present an important binding energy score. Further investigation shows that a range of 156 drug candidates was tiered according to their binding energy scores for each protein. The top three ranked phenolic compounds were subject of residue/molecules moieties interaction analyses. The interaction results revealed that the active sites of each enzyme were conserved with three common residues at least. The stereochemistry of complexes analysed by Ramachandran plot proved that ligands pose do not affect the native enzyme structure. The section of docking screening gave nine phenolic compounds as a potential SARS-Cov-2 inhibitor which are classified as following: 4 biflavones (Amentoflavone, Sciadopitysin, Bilobetin, Ginkgetin and Dieckol) and 5 tannins (Punicalagin, Tellimagrandin I, Tannic acid and Chebulagic acid). A Drug-likeness analysis was realized to evaluate the ability of these candidates to be a recognized as drugs against covid-19. Only the flavone class shows efficiency by one violation of Lipinski’s rules of five, which in turn is consistent with similar results observed for medicinal chemistry by PAINS and Brenk models. Clearly, all observations of this study point to a further required works in order to examine deeply the possibility of using these molecules, which could be subjected for several pre-clinical studies.